Return to Medical Topics Home Page Cyclodextrin Therapy to
Reverse Atherosclerotic Vascular Symptoms and Attenuate Plaque Burden
Return to CHC Home
Page
Dr. Roberts Updated Cyclodextrin Presentation -12/24
Dr. Roberts' Initial ('23) Cyclodextrin
Presentation
Dr. Ablow
(12/24)
interviews Dr. Roberts about Cyclodextrin
Overview
Cyclodextrin Treatment Options and the FDA Cavadex Embargo
Direct
Angiographic Evidence of Coronary Disease Reversal
CT Angiographic
Evidence of Coronary Disease Reversal
Coronary Artery Decalcification
AtheroCare Cyclodextrin Coronary Artery Calcification Reversal Pilot Study
IMT Stabilization with
Plaque Regression
Turning the Atherosclerotic Ship Around with Lipid Reduction and Cyclodextrin
Cyclodextrin for IMT and Plaque Control when
Cholesterol Control is Not Possible
Cyclodextrin
in Cerebrovascular Disease
Cyclodextrin
Controls Hypertension in Renal Artery Stenosis
Cyclodextrin
Rapidly
Improves Endothelial Function and Vascular Symptoms and Assists in Stroke
Recovery
Cyclodextrin
for Post-EECP Refractory Angina
Cyclodextrin
in Heart Failure
Physician DePlaque Thyself!
Cyclodextrin
Rapidly
Restores Vascular Biology in Angina due to Graft Failure
Cyclodextrin to Improve Endothelial Function and Lower BNP
Overview
Cyclodextrin pulls cholesterol (cell membrane,
lipid droplet, crystalline, and oxycholesterol) out of the vessel wall, in a
process that elaborates nitric oxide (vasodilatory and atheroprotective) and
decreases release of inflammatory cytokines. Your symptoms begin to
attenuate in 2-4 weeks (biochemical effects). With longer use, anatomic plaque
regression can occur (as evidenced below). This molecule will save
millions of lives, decrease our need for vascular procedures, and save billions
of health care dollars.
Within our paradigm, that of Integrative Cardiology,
the focus is risk factor reduction, not to directly reverse anatomic
atherosclerosis, but rather to prevent plaque progression or plaque activation –
and thus protect you from
clinical events (heart attack, angina, and heart failure).
Our innate physiology does provide for Reverse
Cholesterol Transport (RCT), the actual removal of lipid material from the
arterial wall. RCT will decrease plaque size and plaque vulnerability. We can
stimulate RCT indirectly with tight risk factor control, and directly with PPC
(Polyenylphosphatidylcholine – discussed in detail on heartfixer.com).
Beta-Hydroxypropyl Cyclodextrin
(which we will refer to as
HPbCD,
Cyclodextrin, or
CD)
is a novel, negligible risk, FDA cleared for human use (but not specifically in
the treatment of vascular disease), and relatively low-cost
approach to actively stimulate RCT, attenuating ischemic symptoms rapidly while
providing a favorable effect (in months, as opposed to years) on plaque size and
extent.
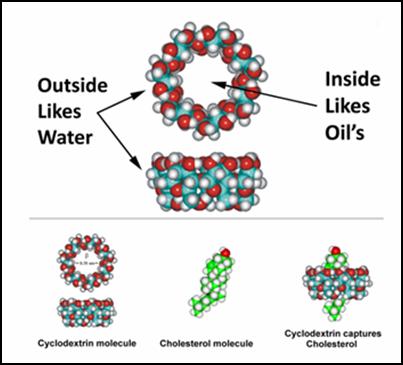 HPbCD,
first administered IV to humans in 1992, consists of seven glucose molecules
bound head-to-tail to form a cone like structure. The exterior of the cone
is water soluble (hydrophilic; thus, it disperses within the blood stream) while
its interior is fat soluble. CD can thus be used (and is), as a drug
delivery vehicle or vaccine component. One molecule of cholesterol fits
tightly within the lipid soluble interior of one CD. If we mixed CD with
cholesterol, the resulting product would transport cholesterol into your cells. Conversely,
as we administer unloaded CD, IV or rectally, we will facilitate egress of
cholesterol (and other undesirable lipid structures) out of the artery wall, and
then out of the body, via the liver (attached to HDL) or out through the kidneys
(attached to CD).
HPbCD,
first administered IV to humans in 1992, consists of seven glucose molecules
bound head-to-tail to form a cone like structure. The exterior of the cone
is water soluble (hydrophilic; thus, it disperses within the blood stream) while
its interior is fat soluble. CD can thus be used (and is), as a drug
delivery vehicle or vaccine component. One molecule of cholesterol fits
tightly within the lipid soluble interior of one CD. If we mixed CD with
cholesterol, the resulting product would transport cholesterol into your cells. Conversely,
as we administer unloaded CD, IV or rectally, we will facilitate egress of
cholesterol (and other undesirable lipid structures) out of the artery wall, and
then out of the body, via the liver (attached to HDL) or out through the kidneys
(attached to CD).
Clearance of cholesterol (into the urine) with
IV CD was described in 1992, but the therapeutic potential of CD was not
exploited until 2010, when CD (IV, sub cut, and intrathecal – into the spinal
fluid) was used for the first time in the treatment of kids with Nieman-Pick.
In this (fortunately rare) autosomal recessive (bad genes from both parents)
condition, Nieman-Pick protein (which functions to shuttle cholesterol from one
cell compartment to another) is defective. Lipids can enter the cell, but
Reverse Cholesterol Transport cannot occur. Liver and spleen lipid engorgement
occurs early in life, with neurologic compromise following soon therefter. CD does not cross the blood brain barrier. Thus, in
Nieman-Pick, this material is infused into the CSF (cerebrospinal fluid), to
treat the brain, with concomitant IV or subcut administration to treat the other
internal organs. Hearing loss typically occurs (a fair trade off), but this
relates only to intrathecal CD delivery; IV or rectal CD will not cross into the
brain and thus will not affect hearing.
So how does this work? Cholesterol can diffuse
out of the cell membrane (a bilayer of cholesterol, fatty acids, enzymes, pumps,
and pores that surrounds our cells) and then attach to a cholesterol receiver
(HDL, albumin, or a red blood cell), but this process occurs ever so slowly.
Cell membrane associated pumps (ABCA1 and ABCG1) and diffusion facilitators
(SRB-1) promote egress of cell membrane cholesterol, but these vascular health
promoting pathways are compromised in patients with atherosclerosis (the problem
is not just too much cholesterol entering the vascular wall, but impaired RCT,
such that Mother Nature cannot rectify the resulting cholesterol imbalance). CD works by
facilitating cell membrane cholesterol egress, upregulating the RCT pumps,
shuttling cholesterol from the cell membrane to HDL (which carries the
cholesterol to the liver) concomitantly diminishing oxidative and inflammatory
stress within the vascular wall.
Further explanation is in order:
A. One molecule of CD can “pluck” one molecule
of cholesterol from the cell membrane surrounding a vascular cell. From there
the cholesterol-loaded CD can be excreted in the urine or CD can transfer its
cargo (a single cholesterol molecule) to HDL (HDL can house 100s of cholesterol
molecules). In this fashion CD acts as a “shuttle” and HDL as a “sink”, a
high-capacity cholesterol acceptor.
B. Excess intracellular cholesterol is stored as
cholesterol ester (cholesterol bound to a fatty acid). Cholesterol ester is
water insoluble; it cannot diffuse out of the cell and creates intracellular
lipid droplets (soft plaque). Acetyl-Cholesterol Acyl Transferase (ACAT),
upregulated by inflammatory mediators, converts free cholesterol (FC) into
cholesterol ester (CE), while Cholesterol Esterase (upregulated by therapeutic
PPC) dissolves CE to liberate free cholesterol (FC) which can then egress the
cell. As CD decreases cell membrane FC, a gradient is created between the
concentration of FC in the cell membrane and that within the cell fluid. ACAT
will be turned down and CE activated. Lipid droplet esterified cholesterol is
thus mobilized to replenish the cell membrane FC pool. With ongoing CD
exposure, intracellular lipid droplet cholesterol overload will resolve.
C. Within the atherosclerotic vascular wall,
cholesterol (particularly oxidized cholesterol) is also stored, within cells and between cells, in crystalline
form. Crystalline cholesterol stimulates plaque inflammation (via activation of
the NLRP3 inflammasome, a plaque destabilizing pathway which we block with
Colchicine - discussed on this website). Foam cells (activated vascular wall
mononuclear white cells, endothelial, and smooth muscle cells) will pinocytose
(“drink in”) CD. The now intracellular CD will dissolve the crystalline
cholesterol, creating a class of compounds termed oxysterols. Oxysterols, in
turn, increase the expression of a class of molecules (ATP binding cassette
proteins ABCA1 and ABCG1) that pump FC out of the cell, making them available
for HDL binding. ABCA1 and ABCG1 promote HDL maturation and enlargement
(here size does matter, as HDL efficiency correlates directly with HDL particle
size). ABCA1 and ABCG1 activation also rapidly turns down inflammatory
cytokine formation. Macrophages (white blood cells within the vascular
wall) are "reprogrammed" from pro-inflammatory foam cell to an inflammation
reduction role (this occurs at other sites of fatty degeneration - such as fatty
liver and recent stroke).
D. At this point CD is pulling, and the
activated cassette proteins are pumping, cholesterol (liberated from their prior
liquid and crystalline states) out of the vascular wall. FC is “handed off” to
HDL, where it is esterified (a process stimulated by therapeutic PPC) followed
by transport to the liver for subsequent GI tract excretion.
E. CD changes vascular anatomy (cholesterol
depletion) and it also changes vascular physiology. The removal of cholesterol
crystals greatly diminishes vascular wall inflammation (like taking
colchicine). CD will also “pluck” diacylglycerols (DAGs) from the cell
membrane. DAGs inhibit Nitric Oxide Synthase (NOS), the enzymes responsible for
NO generation and thus endothelial tone. Endothelial function improves, leading
to vasodilation and (in the setting of occlusive vascular disease) collateral
formation.
F. CD stimulates lysosomal function.
Lysosomes are intracellular digestive and recycling centers, membrane bound
structures containing "hydrolytic" enzymes that break down aged or dysfunctional
intracellular structures (protein, lipids, DNA/RNA) and extracellular molecules
that enter the cell via cell membrane receptors (LDL cholesterol enters via the
LDL receptor) or via pinocytosis (such as cyclodextrin). Cyclodextrin is
just the right size (nanoparticle) to stimulate the production of lysosomes and
their hydrolytic activity. Oxidized LDL, the initiator of atherosclerosis,
poisons the lysosome. Oxidized LDL thus cannot be processed to
free cholesterol; it hangs up within the poisoned lysosome, and then crystallizes
(crystalline cholesterol is far more problematic than lipid droplet esterified
cholesterol). Cyclodextrin rights this metabolic wrong, accelerating lysosomal
degradation of crystalline cholesterol. Autophagy, lysosomal recycling of
aged or dysfunctional structures, declines with age and disease states. Thus we
cannot "clear out the dead wood", compromising cellular (and overall) health.
Many principles of anti-aging medicine (rapamycin, intermittent fasting, caloric
restriction), are designed to stimulates autophagy - we can now add
HPbCD
to this list. Thus cyclodextrin is
going to provide benefits above and beyond reverse cholesterol transport and
plaque regression. Many patients relate that they "just feel better" with
cyclodextrin.
G.
In animal models, CD reduces plaque size and
plaque inflammation. Recovery from ischemic insult is enhanced (CD will remove
pro-inflammatory lipid debris from the site of injury). Fatty liver will improve. CD does not inhibit LDL generation, but
as fatty liver attenuates, liver inflammation and cholesterol generation may
likewise decline.
CD can be administered IV (6 gm. infused over 30
minutes; I receive this during our lunch break), or rectally, as a small
retention enema, 4-16 gm/dose, taken
over 30-90 days. I first learned about CD in 7 /22 (from a patient, as
usual). Intrigued, I read every basic science paper I could find (as far back as
1992), and made contact with the
cyclodextrin
team. The first person
treated was myself (I couldn't tell that the IV CD was even going in), and then we
began rectal CD therapy in a group of patients with prior heart attack and
persistent angina on the basis of failed revascularization (their backs were “up
against the wall”). These long-standing angina patients reported symptomatic
benefit in 2-4 weeks. Thus, 2-3 months of CD makes sense for all of our patients
with symptomatic atherosclerotic vascular insufficiency.
This positive early experience, mirroring the
effect of CD in animal models, has led to my recommendation that all of you with
moderate plaque buildup consider a 1–2-month CD program. You won’t feel better,
but your plaque volume and plaque inflammation will fall, a preemptive attack on
atherosclerosis that developed in the past and which may advance despite out
best preventive efforts. Indeed, risk factor control is problematic for many of
you (you can’t tolerate lipid-lowering therapy or there are cost constraints).
CD is well tolerated, safe (side effects are nuisance in nature related to
rectal delivery), and cost is not excessive. In a sense CD is
the universal antidote for atherosclerosis. My thinking here is that 1-2
months of CD every year will “make up” for any imperfections in our risk factor
control efforts.
IV CD is administered via a butterfly needle,
and the infusion runs in over 30 min. IV CD stays in your circulation over six
hours. Thus, if the goal is rapid reversal of atherosclerosis, IV CD could be
administered every 8-12 hours. Cost here would be excessive and thus daily rectal CD makes more sense.
PPC will synergize with CD. We provide PPC IV,
and it can be taken orally (as PhosChol or Plaquex, or mixed with EDTA as
DeToxMax). I take one tsp. of PhosChol (2,700 mg) daily.
This is a great idea, and its widespread
application will help millions of Americans and save billions of health care
dollars. As CD is not patentable, formal research to “prove” efficacy may
never be done. Thus, CD will never be covered by insurance and standard
Medicine will ignore its potential (we won't). The questions that arise as
I advise individual patients about cyclodextrin are:
A. How long should a course of cyclodextrin last?
B. Is maintenance therapy important?
C. At what time interval should we repeat a course of CD therapy?
C. How can we measure efficacy?
The ongoing results of CD therapy in my personal patients is helping me to
answer these questions.
Right now,
given the safety, low cost, and obvious efficacy of CD, my recommendation is:
A. In the setting of symptomatic atherosclerosis, 2-3 months of CD makes sense
(or until symptoms resolve) followed by a maintenance program of 1-2 doses per
week, with repeat 30-day programs once or twice a year.
B. In asymptomatic patients with moderate plaque (particularly if risk factor
control is difficult) one month of CD twice a year can be used to make up for
imperfections in our preventive program.
C. Keeping in mind that CD does not remove the underlying causes of
atherosclerosis, ongoing risk factor reduction remains important, but with the
benefits of CD, we may be able to cut back on the drugs which are most
problematic to you.
D. We can use change in carotid plaque and intima-media thickness (discussed
elsewhere on this site) and coronary artery calcium score, as means of
quantifying CD efficacy.
James C. Roberts MD FACC FAARFM
1/24/25
Cyclodextrin Treatment Options and the Cavadex FDA Embargo
I want
Beta-Hydroxypropyl Cyclodextrin
(HPbCD)
to be available to all of you, ideally with a means of administration that you
can tolerate and afford. Our early experience involved the use of Cavadex HPbCD,
a low volume rectal retention enema. The clinical outcomes presented on
this site all relate to the use of Cavadex HPbCD.
The FDA embargo on Cavadex HPbCD
put all of us in a difficult situation (particularly those of you
who were getting better, only to experience recurrent symptoms when our
government cut off your Cavadex supply)! I communicated with the FDA, verbally
and in writing. I presented patient case studies, demonstrating benefit
of Cavadex HPbCD,
with respect to both symptom attenuation and markers of plaque burden in my
patients.
I pointed out that multiple FDA-approved prescription drugs contain HPbCD
(to improve drug bioavailability), that
FDA has designated HPbCD
as GRAS (generally regarded as safe), based on
multiple safety studies (mostly in animals but some in humans) carried out by
the pharmaceutical industry twenty year ago (I've read these studies - its
really hard to hurt an animal with HPbCD
- your need to administer a ridiculous dose), and that FDA has cleared HPbCD
(in this capacity as an "orphan drug") in the treatment of kids with the genomic
Nieman-Pick lipid lipid retention disorder, and that we as citizens can buy HPbCD
over-the-counter by the truck load. My entreaties (and I put a lot or work into this) had no
effect, other than that one patient (with inoperable and recurrent coronary
disease) was granted an individual embargo exemption.
FDA's position is that
Cholrem presents Cavadex HPbCD
as a "new drug", as Cholrem states that Cavadex HPbCD
can reverse specific disease symptoms, lower lipid values, and decrease plaque
volume. They object to
Cholrem presenting, on their website, my words, writings, and audio-visual
presentations that support this position. The FDA has no problem
with me, as a Doctor, saying whatever I want, but that my evidence or position
statements can't be used in the marketing of substances with "new drug" claims.
The FDA officials who I spoke with (nice people by the way) related that
non-drug agents can be marketed to support various aspects of health,
but that
if a disease mitigation claim is made, that this must be backed up by a FDA
sanctioned study. Thus when you buy a supplement the label will not say "lowers
cholesterol" or "lowers blood pressure". Rather that label
will say
something more general such as "supports cardiovascular health". I
get this, in that unsavory individuals could make things up and trick desperate
people into buying ineffective or dangerous substances. On the other
hand,
I am not an unsavory person. I want the world to know about the myriad benefits
of HPbCD.
My message to FDA is that widespread utilization of
HPbCD
will decrease your need for invasive procedures, save million of
lives, and billions of Medicare dollars! No one was interested (but you are)!
A path forward, if Cholrem wanted to
market Cavadex HPbCD
within this "new drug" format, would be to spend tens of millions of dollars
proving to FDA what we already know, that HPbCD
is safe and effective in the
treatment of vascular disease. At that time, Cholrem could market their product
in the US, with drug claims, but so could I, so could you, and so could anyone else.
Cholrem would never recoup their investment. Such research
will thus never
be
done (but Medicare will cover the cost of repetitive hospitalizations and
invasive procedures). We had initiated a fifty patients, open study, to assess
the effects of six months of Cavadex HPbCD
on coronary calcium score
findings. Cholrem was going to supply the HPbCD
and cover the costs of the calcium scoring. This informal "pilot study" would not be accepted by
FDA as proof, but it
would have provided a great deal of
information as to how we can best use HPbCD.
Of course, with the FDA embargo, we shut this study down.
FDA wrote to Cholrem,
apprising Cholrem of their concerns, with specific, line-by-line,
directives as to what their website could not say (I was to be banished)!
Cholrem refused to do so. Like me, Cholrem wants the
whole world to know about the benefits of HPbCD.
As Cholrem provide Cavadex HPbCD
to individuals around the world, in countries that do not have this restrictive
policy, they did not want to "water down" their
position. Cholrem dug in its heels, and FDA began to embargo and destroy
incoming Cavadex HPbCD
shipments. You were left in the medical lurch, and Cholrem sustained a
massive financial hit (for trying to help you)!
I have no financial entanglement with
Cholrem nor any other HPbCD
manufacturer, but as you can tell, I am "all in" on this concept, and have
certainly been in communication with Cholrem. While I understand
their position, I have never found that "digging in your heals" against the US
government to be a successful long-term strategy, particularly if you can modify
your position such that you can achieve your goals without government
interference. I am hopeful that Cholrem will modify their website and marketing
materials, such that shipments of Cavadex HPbCD
into the US can be resumed.
In the meantime, we
can't let this regulatory snafu set your back, and in relation to the FDA
Cavadex embargo, three alternative sources of HPbCD
have been brought to my attention, as presented below:
Atherocare, like Cholrem, based in Australia, provides HPbCD
within the same small volume retention enema format. This is the
same stuff. I recently spoke with a physician (on the other side of the Equator)
who, after
viewing my initial HPbCD
presentation, began a one year program of Atherocare, along with his own best
anti-atherosclerosis treatments. He had presented with new onset symptoms and
two vessel (one 98% and one 90% obstructed) coronary disease. Stent
placement could not be carried out for three months (yes, our system has
problems, but can you imaging waiting three months for a stent)? By the
time they called him in, he was feeling better.
This Doctor skipped the
stent, and now at the one year point his symptoms are minimal. He called me to
let me know and he invited me to his home (he lives on a mountain overlooking the
Amazon basin).
Several new patients
had learned about
Atherocare and had obviously benefited from its use. Thus, at this time, we are using
Atherocare just as we used Cavadex. Atherocare, in response to the FDA
guidance letter, scrubbed their website of all
statements that could be
construed
as a disease mitigation claim. They dropped my audio-visual presentations (I seem to be getting banished a
lot these days)! As a result, FDA is not embargoing Atherocare. Atherocare
is
working within FDA guide lines. They are not marketing their product.
They do not need to. Word of mouth, and the enthusiastic support of
integrative providers such as myself, is going to get the word out for them!
This policy
is working out for all parties
Oral
HPbCD
has demonstrating some anti-atherosclerotic efficacy in animal studies, but
in us humans only 3% is absorbed. The rest may bind up fatty materials and
cholesterol (and thus may lower your lipid values) and the
cyclodextrin-cholesterol complex is then conducted to the large intestine.
This fat burden can lead to rectal fatty leakage (stains on your underwear), and
the cyclodextrin molecules can be fermented by GI microbes, creating
useful metabolic intermediates (runners in Japan do better when on oral
cyclodextrin) but then copious intestinal gas is created. As I always try
new treatments out on myself, I took in two teaspoons of oral HPbCD,
and soon
found myself in a situation where I needed to avoid others the rest of the day. Oral HPbCD
is thus not the way to go!
HPbCD
Troches: In relation to the FDA Cavadex embargo, I asked Kieu Okuley
RPH, of Okuley's Compounding Pharmacy, in Defiance Ohio, to create a non-rectal
HPbCD.
Kieu did the research and came up with 1000 mg
HPbCD
troches. These semi-solid cubes, placed between your cheek and gum, will
dissolve slowly, such that the HPbCD
is taken up by the oral capillary microcirculation, as opposed to swallowed into
the GI tract. I have
taken the troches twice a day without GI distress. How HPbCD
troches will compare to rectal HPbCD
is a great question that I will try to answer, based upon how my patients
respond. HPbCD
troches can be obtained from
Okuley's with a physician prescription. While the HPbCD
troches dose is fixed at 1000 mg, it is up to us as to how many troches to take
per day (they take 20 minutes to dissolve).
Albedextrin formulated by
Spencer Feldman of remedylink (remedylink.com)
is a blend of alpha and beta cyclodextrin, designed for
oral use. I learned about Albedextrin from a patient (like everything else I do
that is good).
Talking with Mr. Feldman is like talking to my organic chemistry professor is
college - Spencer knows his formulation chemistry! I will not give away the
secret, but Spencer found a way to enhance cyclodextrin absorption. Albedextrin
is provided in powder form. You mix one pouch with water, store it in the
fridge, and take a dose on a regular schedule (instructions on the
remedylink.com website), cutting back on the dose or frequency if
nuisance GI side-effects occur. As oral cyclodextrin can bind up
phosphatidylcholine and fat-soluble vitamins, Spencer recommends corresponding
supplementation if long-term use of Albedextrin is planned. Spencer has a
number of other quite interesting supplements, several of which I am now taking.
His website provides a plethora of information. As with cyclodextrin troches,
over I time I will develop a feel as to how these new cyclodextrin approaches
compare to rectal
HPbCD,
the benefits of which are documented elsewhere on this web page..
James C. Roberts MD FACC FAARFM
7/20/25
Direct
Angiographic Evidence of Coronary Disease Reversal
If you benefit from cyclodextrin, then thank Kyle Hodgetts. Kyle, a
hypertensive, hyperlipidemic, long-standing smoker, by age 56, had required
several rounds of stent placement. Standard medicine was not working.
Kyle's physicians gave him little hope for future health. The adage
"Necessity is the Mother of Invention" certainly applied here. Kyle had
just lost his wife to illness. The couple had two young children. What would
happen to them if Kyle died? Kyle, a researcher, read Dr. Zimmer's article
(convincing animal model evidence that cyclodextrin will reverse
atherosclerosis). I also read this article too, got excited, and soon after lost
interest as cyclodextrin was then not available in the US. Kyle read the
Zimmer study, and then every other paper he could find on this molecule (there
is a lot - I've now read it all too). Kyle found a source of pharmaceutical
grade CD, and then, assisted by his medical and scientific team, began to treat
himself with IV cyclodextrin. Kyle and his medical group kept careful
records and published the results. As depicted below, Kyle experience rapid
(months, not years) coronary disease regression (documented by angiography),
corroborated by carotid plaque regression (documented by ultrasound), and his
lipid panel improved. Representative images are presented below. You can
read the published reports yourself (Kyle's
coronary angiogram, Kyle's'
carotid ultrasound). Kyle tells his story, and discusses the science
of cyclodextrin, on cholrem.com. You will
note that everything Kyle says is pretty much everything that I am saying.
I am not parroting Kyle, nor visa versa. We both studied the literature and came
to the same scientific conclusions. The outcomes experienced by Kyle's clients
are just what my patients are experiencing (as presented below).
Cyclodextrin, once it is understood by all (a job I have assigned to myself) and
put into widespread use, is going to save millions of lives, decrease the need
for surgical interventions, and save billions of dollars. Kyle, in this regard,
deserves a Noble Prize - he brought this to you!



CT Angiographic
Evidence of Coronary Disease Reversal
Coronary angiography (injecting X-ray contrast dye into the arteries serving the
heart) is the "gold standard" in coronary imaging, but carries with it risk. I
know about this as I have carried our 1000s of them. The risk of death is 1 in
1000 and the risk of a complication is 1 in 100. Thus we carry out invasive
angiography only in situations where revascularization will likely be necessary,
or when a question exists that cannot be answered with non-invasive testing -
this is always a risk vs. benefit decision.
CT angiography (CTA) involves much less risk. Here we inject contrast dye into a
peripheral vein, followed by CT rapid imaging of the vessels of interest.
Image quality is not as crisp as that afforded by direct arterial injection, but
CTA does allow for reasonably accurate quantification of disease burden.
Stated otherwise, CTA can answer most of our questions, and can do so with low
risk. CTA is often combined with coronary artery calcium scoring (CAC),
which we will discuss in the next section.
CC, a then 63 year-old women with statin intolerant hyperlipidemia and elevated
Lp(a), underwent CTA in 4/22 to evaluate effort-related chest tightness.
CC's CAC was 212 (90th percentile for age) and a 70% LAD (left anterior
descending, which serves the front wall of the heart) narrowing was identified.
CC's symptoms were not unstable, and a 8:30 stress echo returned benign.
Thus anti-ischemic medical therapy was initiated, with a plan to proceed with
invasive angiography and stent placement if CC's symptoms did not resolve.
CC was also troubled by chronic GI symptoms, including SIBO (small intestinal
bacterial overgrowth), H. pylori gastritis,, yeast overgrowth, and food
sensitivity. CTA imaging also suggested impaired arterial flow to the GI tract.
CC had a great deal of trouble simply maintaining her weight.
Standard medical therapy was poorly tolerated. We began working together in
9/22. CC's program included Lp(a) binding inhibition therapy and Lumbrokinase
(aiming to prevent plaque progression) along with phosphatidylcholine and
cyclodextrin (to promote reverse cholesterol transport). This program
worked. Three months later CC's angina had all but resolved. CC's CTA was
repeated in 8/23, following a total of 120 doses of cyclodextrin, and described
a mild-to-moderate LAD narrowing. Cleerly secondary analysis of the data
described 27% LAD, 12% circumflex, and 8% right coronary narrowings (Clearly
allows quantification of plaque extending into the artery wall as well as that
of flow-limiting potential). Through 1/25, with weekly maintenance
cyclodextrin. CC remains asymptomatic, her GI situation has improved, and CC is
getting on with life.
In CC's case, rather than carrying out revascularization of her LAD (and
possibly her GI tract arterial supply), putting stents in to "lesions" (our term
for loci of dysfunction) we instead treated her "circulesion", the phenomena of
atherosclerosis per say. Our approach is better, you are waking up to it
(if not you would not be here), and some day insurers will as well.
As CC's CTA exams were carried out on different machines and read by different
doctors, a non "apples to apples" comparison, going from 70% to 27% is not a
trick of the eye. Our initial pilot study was to be a CAC assessment in
fifty patients (see below), but the next logical step would be a CTA assessment
of cyclodextrin efficacy (right now government is working against us, but some
day they will be working with us).
Coronary Artery Decalcification
In relation to age,
Vitamin K2 status (I take 1000 mcg each day),
and more importantly, to the extent and chronicity of one's atherosclerotic burden,
arteries will calcify. This can be quantified with a simple, low cost
($100), relatively low dose (equivalent to one years worth of environmental
radiation exposure) X-ray study, the Coronary Artery Calcium Score (CAC).
CAC relates to disease burden, and less precisely to age-related risk.
The CAC report will provide a total CAC,
along with a scores for each individual artery. The data will also be expressed
as an age-related percentile (how you compare to your age mates). A
calcium score of 100 in an 80 year-old would be relatively low, and not
associated with risk, while the same score in a 30 year old would be well
above the 95th percentile, reflected premature disease, with a heightened event risk.
Other diagnostic studies, such as EndoPAT
endothelial function assessment and Carotid Artery IMT (Intima-Media Thickness), reflect recent vascular
biology, such that we can use change in these parameters to assess the efficacy of
our preventive efforts. In contrast , your CAC reflects the life-long
build up of atherosclerotic plaque, and not your recent corrective efforts.
However, change in CAC, as with change in IMT or change in endothelial function, can be used to assess treatment efficacy and gauge event
risk.
 In
the average American, CAC progresses at 20% per year (that doesn't mean that
your arteries are narrowing at 20% per year, rather that arterial calcification,
a surrogate for atherosclerosis, is progressing at this level). Yearly CAC
progression above 20% is associated with a heightened event risk (the greater
the rate of CAC progression above 20% the greater will be your risk), while CAC
progression at ≤ 15% is associated with far lower risk (things are under
control and calcification is no longer progressing rapidly)!
In
the average American, CAC progresses at 20% per year (that doesn't mean that
your arteries are narrowing at 20% per year, rather that arterial calcification,
a surrogate for atherosclerosis, is progressing at this level). Yearly CAC
progression above 20% is associated with a heightened event risk (the greater
the rate of CAC progression above 20% the greater will be your risk), while CAC
progression at ≤ 15% is associated with far lower risk (things are under
control and calcification is no longer progressing rapidly)!
A younger individual with a CAC of 100 and yearly progression at 30% is more
likely to get into trouble than is an older individual with a CAC of 1000 with
10% progression.
Many of our therapies have been shown to delay disease progression and protect
against clinical events, but none have been shown to halt, let alone reverse, CAC
progression. Statin drugs have many benefits, but as they deplete the body of
Vitamin K2, these agents may actually accelerate CAC progression (we can compensate
here with K2 supplementation).
Our patients are certainly benefiting from cyclodextrin, and as your will see
below, we have documented carotid bulb soft plaque regression, but why would
cyclodextrin affect CAC, which we view as an indicator of "hard plaque"?
On the other hand, the mechanisms of vascular calcium build up are not well
defined. We know that cyclodextrin will dissolve crystalline cholesterol as well
as intracellular lipid droplet cholesterol.
So what affect will cyclodextrin exert on the rate of CAC progression?

RJ, a 75 year-old, physically active man with chronic atrial fibrillation and
elevated Lp(a), is not my patient (a family member is). RJ forwarded his
history and CAC findings, and graciously allowed me to share his cyclodextrin
CAC response with you.
RJ's CAC had progressed from 783 in Nov. '15 to 1405 in March '23; CAC
progression at 24% per year. RJ learned about cyclodextrin, and over the summer
of '24, RJ completed three months of daily treatment (with the 15 gm dose),
followed by repeat CAC testing that Sept. RJ's CAC, which has been
progressing at 23% per year, had now regressed by 8% (6% per year from March
'23, an underestimate of treatment effect as CAC had been progressing from
March '23 until RJ began cyclodextrin therapy 1 1/3 years later in mid '24).
RJ's Nov. '15 CAC was not stratified artery-by-artery, but his latter two CAC
studies were, and as you can see, CAC regression occurred across the arterial
board. To me, this
is unprecedented!

BP works with a close colleague who follows his patients' coronary status with
serial CAC testing, utilizing CAC rate of change as an indicator of treatment
efficacy and adverse event risk. BP watches his diet and works out on a
regular basis, but this 59 year-old man has been challenged with
hypertension, hyperlipidemia, elevated Lp(a), and a host of other risk
factors.
BP's CAC regressed a point between Aug. '16 and Sept. '18, but then skyrocketed
by
636% (127% per year), over the ensuing
five year period. BP received five months of CD, spring-summer '24, and then
repeated his CAC. Just as with RJ, BP's CAC, which had been progressing
rapidly, had now regressed by 6%.
PA
bears pronounced, mixed hyperlipidemia (likely inherited from his Dad, who
sustained a heart attack at age 42). We began working together in mid
''22. At that time PA's CAC was 72, and ultrasound revealed a moderate carotid
bulb plaque burden (all reflecting the lifelong effect of PA's risk factors).
Conversely, endothelial function was intact (EndoPAT score of 1.79), and PA's
mean IMT, which along with endothelial function reflects more
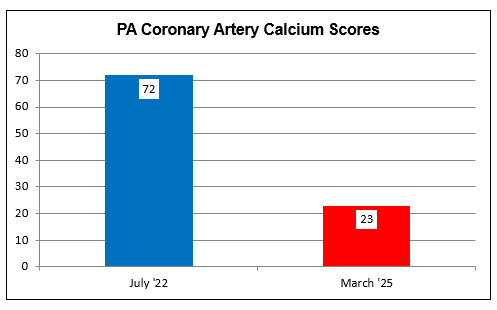 recent vascular
biology, was actually below-average-for-age at 0.711 mm. This discrepancy
between past (CAC and plaque burden) and present (low IMT and preserved
endothelial function) reflects PA's comprehensive program of nutritional
supplementation (which included K2) and PA 's favorable diet and lifestyle
practices.
recent vascular
biology, was actually below-average-for-age at 0.711 mm. This discrepancy
between past (CAC and plaque burden) and present (low IMT and preserved
endothelial function) reflects PA's comprehensive program of nutritional
supplementation (which included K2) and PA 's favorable diet and lifestyle
practices.
We added lysine and proline, aiming to blunt Lp(a) trapping within the arterial
wall, and Jiagolan to lower TMAO. PA strongly did not wish to receive statin
lipid-lowering therapy (and with these favorable vascular biology findings I did
not need to arm twist), but we were able to lower PA's LDL form 259 mg/dl to a
nadir of 167, later rising to 200, with a well tolerated program of RYRE and
Berberine - not great risk factor control but we were making progress.
To cover for atherosclerotic progression that might be breaking through PA's
anti-atherosclerotic risk factor reduction program, PA completed two months of
cyclodextrin therapy in '23 with a second two months program in '24. Thus we
were taking direct aim at atherosclerotic plaque build up with cyclodextrin
while dong our best to control PA's atherosclerotic risk factors, utilizing
therapies that he could tolerate.
To assess efficacy we repeated PA's baseline studies and observed
an across-the-board improvement. Endothelial function, OK at baseline, had
improved further (EndoPAT score rising from 1.79 to 1.99). Carotid IMT had
regressed from 0.711 to 0.704 mm), and PA's CAC had decreased by 68%, or 25% per
year, from 72 in 7/22 to 23 in 3/25 (please remember that the average American
demonstrates CAC progression at + 20% per year and IMT progression at 0.01
mm/year). Of course, PA is not an average American. Rather PA is a
thoughtful, proactive individual who is taking charge of his vascular health. PA
is going to continue his current program, which will include two months of
cyclodextrin every year, with a plan to monitor lab studies periodically and to
repeat PA's IMT study at the one year point.
SR bears pronounced, mixed hyperlipidemia, with a peak LDL of 202 mg/dl in 2/22,
and Lp(a) of 155 mg/dl (reference limit 75), identified in 2/23. While
asymptomatic, SR's CAC had increased by 23% per year, from 84 in 9/19 (7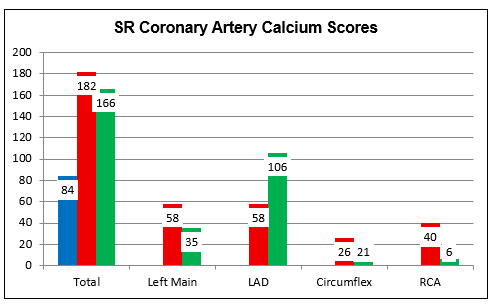 5th to
90th percentile for age), to 182 (89th percentile for age) in 7/23. Carotid
ultrasound in 8/22 describe Right Internal Carotid Artery flow-reversal,
suggesting a critical narrowing. This led to CT angiography in 1/23 which
described an 86% narrowing.
5th to
90th percentile for age), to 182 (89th percentile for age) in 7/23. Carotid
ultrasound in 8/22 describe Right Internal Carotid Artery flow-reversal,
suggesting a critical narrowing. This led to CT angiography in 1/23 which
described an 86% narrowing.
SR and I began working together in 10/23. Lp(a) binding inhibition therapy with
lysine and proline was added. SR strongly did not wish to take statin drug, but
with a programs of RYE 6,400 mg, Berberine 1000 mg, and Niacin 1000 mg/day,
LDL fell to 169 in early '25.
In late '23 SR began a five months of nearly daily cyclodextrin, followed by
maintenance therapy with one dose (double strength) each week, while continuing
her prior, quite comprehensive, nutritional program and pristine diet.
SR's carotid artery CT angiogram was repeated in 4/24,
demonstrating an 80% RICA narrowing (down from 86% in 1/23). Her calcium score
in 5/25 returned at 166 (84th percentile for age), regressed by 5% per year
from 7/23, with long-term progression from 9/19 at 17% per year.
SR is going to continue weekly cyclodextrin, along with her other treatments,
with periodic lab monitoring an repeat non-invasive studies in two years.
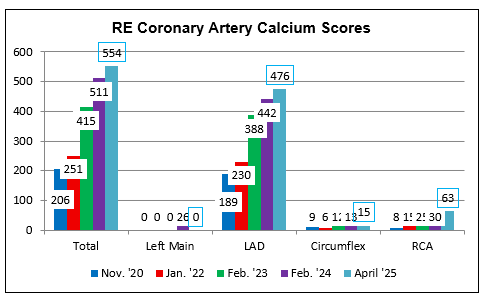
Dr. RE is not a fan of the Fauch. Nonetheless RE was
able to "flatten the calcium score curve" with cyclodextrin.
RE experienced new onset of atrial
fib developed in 2/20, related to an inadvertent change in RE's medical regimen.
Echo demonstrated mild pump dysfunction (ejection fraction 35-40%),
moderate-to-severe mitral regurgitaon, and thrombus (clot) within RE's left
atrial appendage. Kidney dysfunction (creatinine 1.4 mg/dl) was noted.
Anticoagulation was initiated followed by medical cardioversion back to normal
rhythm. Pump function normalized and mitral regurgitation resolved. RE's CAC returned at 206 (70% percentile for age); concomitant CT
angiography described a 50% mid-LAD narrowing.
CAC scoring was repeated a little over one year later and returned at 251, an
18% per year increase (as discussed above, outcome relates more to rate of
change in CAC as opposed to the absolute CAC value). The LAD narrowing was
stable at 50%.
RE and I began working together in 5/22. Vitamin K2 was added to help blunt CAC,
Allopurinol to blunt oxidative stress and to protect RE's kidneys, (creatinine
is now in the 1.2s), homocysteine dropped from 17 to 12, and a LDL of 45 mg/dl
was achieved. We carried out our usual maneuvers but were distressed to see a
65% per year rise in CAC to 415, as of 2/23, with an increase in LAD stenosis
into the 60-75% range.
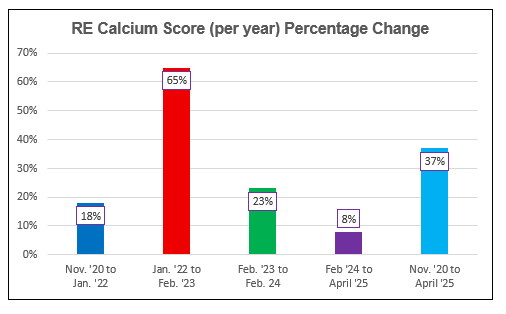
To RE's program we added two months of cyclodextrin in '23, and with this RE's
CAC increased only 23% per year, 2/23 to 2/24, with one additional month of
cyclodextrin in '24, and with this RE's CAC yearly, now with a stable 70-80% LAD
narrowing.
RE does experience shortness of breath with brisk walking, but not with weight
training. We understand that in the presence of vascular calcification, that %
narrowing on CT angiography can be an over-estimate. RE's plan is to complete
two additional months of cyclodextrin, and then to undergo perfusion imaging, to
help us determine whether the LAD narrowing is or is not flow restrictive.
With these positive findings, we initiated a 50
patient pilot CAC regression study, but had to stop in relation to the FDA
embargo. Hopefully the politics/regulations will change with the new American
Administration so we can get back to helping people with this low-cost,
low-risk, non-invasive approach.
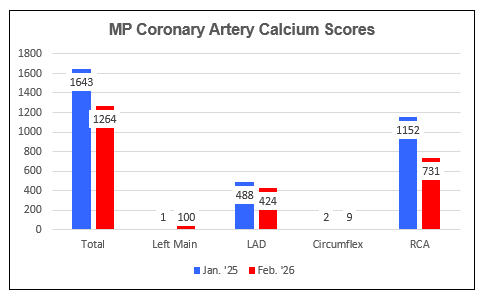
MP's Dad required bypass surgery at age 60 and here MP is uninterested in
following in his Father's footsteps. Thus when screening CAC scoring in
1/25 returned at the 90th percentile at 1,643, MP and his physician initiated
corrective action.
Berberine and Ezetimibe were added to prior statin therapy and with this LDL
fell to 59 with an HDL of 56. 2000 mcg/day of K2 was added to MP's already
comprehensive nutritional program. MP received 12 IV EDTA and 12 IV Cyclodextrin
infusions, and in 5/25 initiated a program of Atherocare Cyclodextrin, twice a
day by three months and then daily over three additional months. MP tolerated
this protocol well.
MP's Calcium Score fell by 424 points, from 1643 to 1264, a 26%/year rate
of regression (this underestimates the AtheroCare effect, as five months had
elapsed between MP's 1/25 study and the initiation of AtheroCAre.
MP is going to keep p with AtheroCare at a maintenance dose of 2-3 doses per
week and we will repeat his CAC study in the spring of '27.
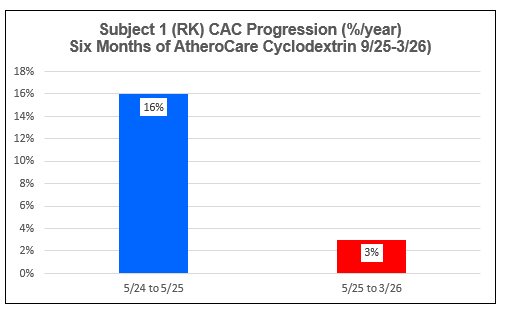
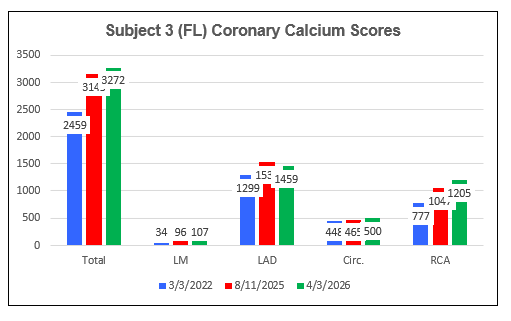
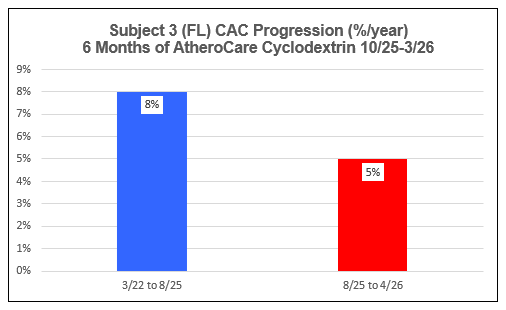
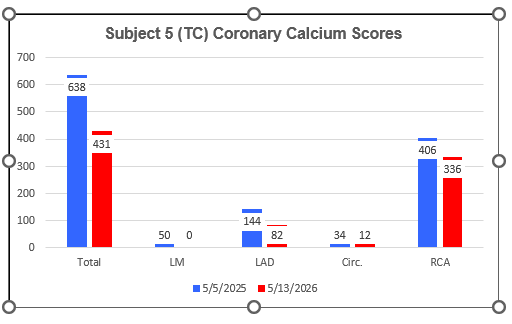
AtheroCare Cyclodextrin Coronary Artery Calcification Reversal Pilot Study
Group Results to Date (7/22/26)
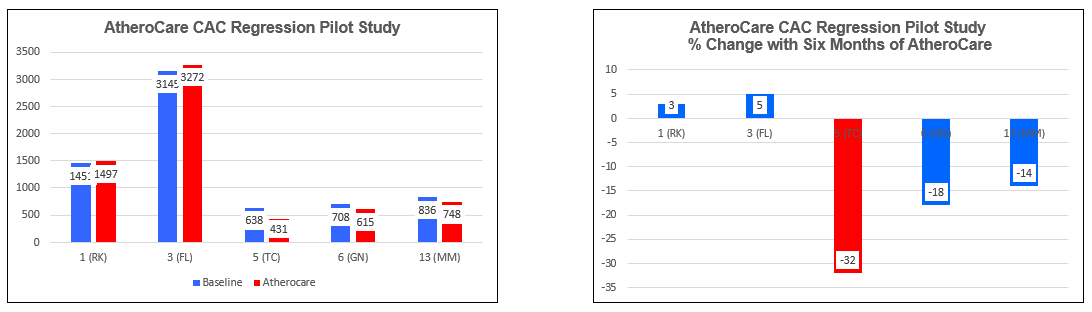
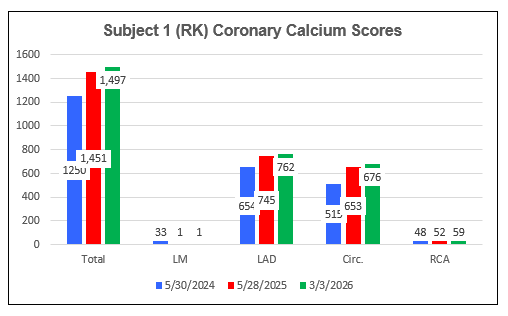
Subject One (FL)
Subject Three (FL)
Subject Five (TC)
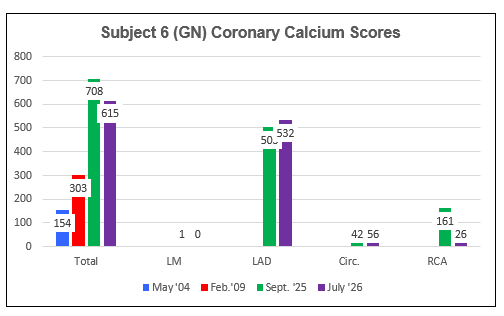
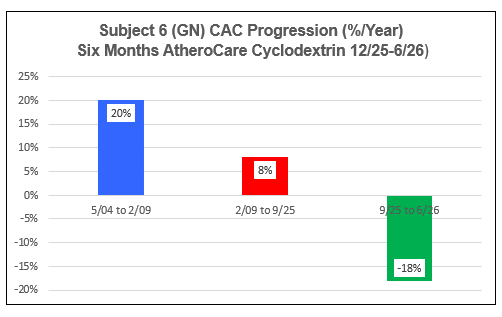
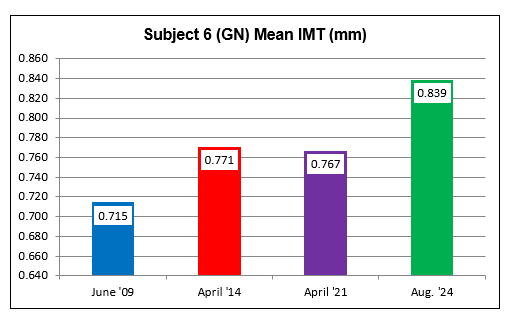
Subject Six (GN)
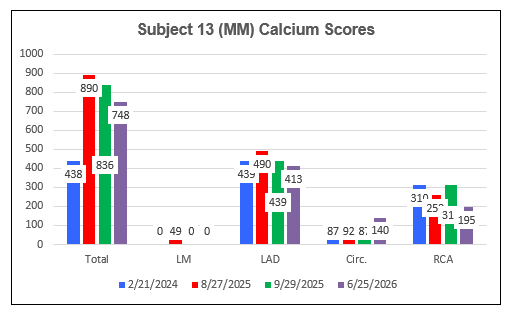
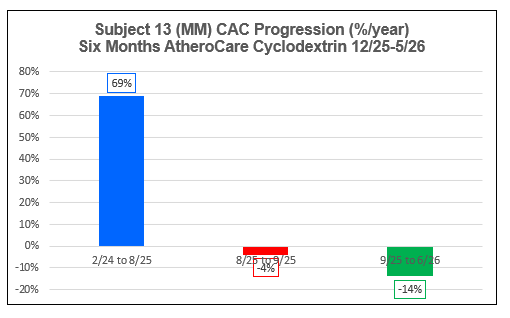
Subject 13 (MM)
IMT Stabilization with Plaque
Regression
Carotid
Intima-Media Thickness testing (CIMT), my favorite measuring stick as to
current vascular health, is discussed in detail elsewhere on this website.
The greater your IMT, and the greater the rate of IMT progression, the more rapidly are
you generating new plaque, and the more likely are you to experience a clinical
event. Conversely, if we stabilize or regress your IMT, then event risk is
low (we are" turning the ship around"). The CIMT study does not visualize the
deeper regions of your internal carotid arteries, nor does it estimate their
degree of narrowing (here we would us the standard carotid ultrasound), but the
CIMT study does visualize the common carotids and carotid bulbs, and here we can
assess changes in plaque burden.
SM, now in her mid-60s, is incredibly health conscious, a good thing in that she
bears genomic hyperlipidemia, with a 2011 baseline LDL of 194 mg/dl. With
RYRE 2,400 mg and Berberine 1,000 mg/day, LDL fell to125 mg/dl, at which time
SM's baseline IMT study was carried out. As plaque was identified, we
switched from RYRE to Rosuvastatin 10 mg/day (keeping Berberine on board), and
with this SM's LDL dropped to 102 in 4/22. Myalgia then occurred, prompting a switch
to Pravastatin 20 mg/day, which didn't work as well, with a LDL of 182 in 12/22.
Given our inability to control SM's hyperlipidemia, SM received two months of CD
therapy, without difficulty. SM's IMT study was then repeated,
demonstrating mean maximal IMT regression, and minimal mean IMT progression (a positive
physiologic sign) and to my eye, a
regression of carotid plaque. Being intellectually honest (I don't work
for the government and thus honesty is expected) the three-year improvement in SM's IMT findings could relate to all of the other, non-lipid reduction
maneuvers that SM has been carrying out, but with these measures alone I rarely if ever see visible plaque
regression. In animal models (and in cases posted on another website) visible plaque regression may occur within months, and
thus this dramatic change in SM's findings likely reflect CD therapy. Our
tentative plan is for SM to complete one months of CD twice a year, with
periodic IMT reassessments.
Baseline Study in 3/20
Post-Cyclodextrin therapy in 2/23
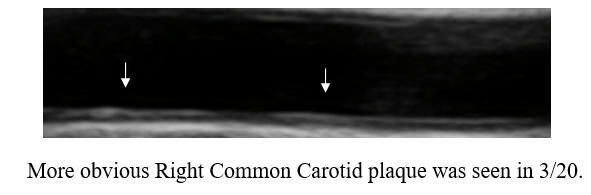
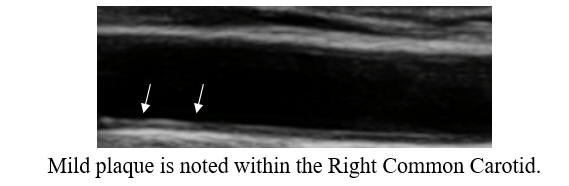
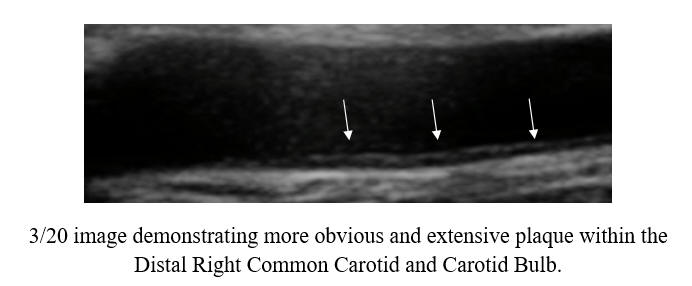
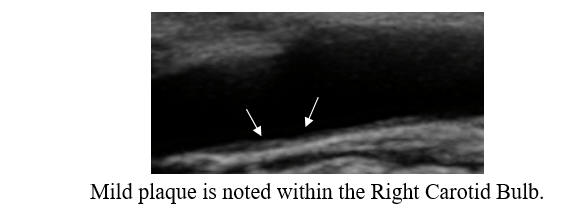
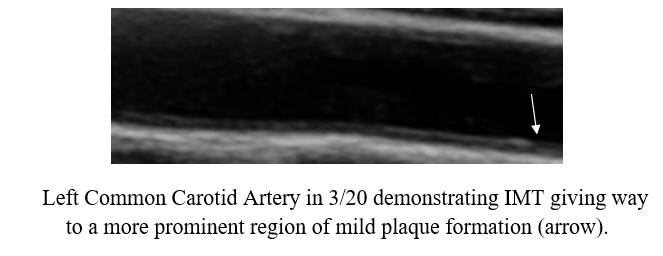
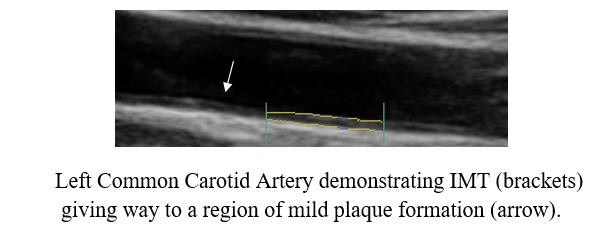
This represents a reversal of IMT (the current status of one's vascular biology)
and a regression of atherosclerosis (that developed over decades), utilizing a
well tolerated, safe, and relatively inexpensive (invasive revascularization
costs a minimum of $10K) therapy. Now, SM's myalgia was later found to be
due to fibromyalgia, which is responding to physical therapy, and thus she is
going to resume Rosuvastatin, which should help in the long-term, and which
should synergize with future rounds of CD therapy. SM's outlook, despite
genomic hyperlipidemia, is certainly positive, now that we have found a means of
reversing her atherosclerosis.
Angina Regression relates to Carotid Plaque Regression
ME, a then a 62 y/o
women with DM II, high Lp(a), and (statin intolerant) hyperlipidemia, was
hospitalized in 5/18 with atypical chest discomfort. Coronary angiography
demonstrated a smooth, 70% LAD narrowing, just on the cusp, with respect to the
need for stent placement. Our usual risk factor reduction maneuvers were
initiated, but medication intolerance precluded tight cholesterol and glucose
control. Still, ME felt well, free of effort-related symptoms. Her 5/20
carotid mean IMT was average-for-age; mild carotid bulb plaque was identified. A
4:30 stress echo study carried out in 2/12 returned normal
A set back occurred in in the spring
of '23, when ME began to experience chest tightness and shortness of breath with
previously well tolerated effort. My interpretation was that ME's 70% LAD
narrowing was now flow restrictive. ME was not enthused about angiography and
stent placement. Cyclodextrin was working well for our other patients.
Thus a trial of cyclodextrin served as a logical first step. After sixty
doses, ME's symptoms had resolved. A 3:31 stress echo study returned
normal, corroborating ME's symptomatic improvement. After 180 doses, we repeated
ME's carotid study, and identified IMT regression (0.685 mm in 5/20, decreasing
to 0.675 in 9/23), coupled with an element of plaque regression. ME
completed 8 months of cyclodextrin through 12/24, at which time she switched to
maintenance program of two doses per week.
ME's symptoms resolved, carotid plaque burden lessened, and our inference here
is that her LAD had opened up enough to restore normal blood flow. Our
plan is to keep ME's LAD (and the rest of her vasculature) open with risk factor
control (as tolerated) and periodic use of cyclodextrin.




IK, then 57 years-of-age, in 7/06 underwent three-vessel bypass surgery.
Health aware and highly motivated, ME put himself on a comprehensive risk factor
reduction program, and in 5/08, ME's mean IMT was actually below average-for-age
and only mild carotid plaque was seen, with little change two years later.
IK developed the great habit of swimming vigorously, three days a week
Effort-related symptoms (swimming became a challenge) recurred in mid-'22. A
stress echo study demonstrated ST depression (suggesting impaired blood flow),
but concomitant stress echo imaging was normal. CT angiography was carried out
to settle this discrepancy. All three of IK's grafts were open, but a 70%
narrowing had developed in the non-bypassed right coronary artery (only 30% in
6/06), likely the culprit. Concomitantly, carotid plaque progression
was identified.
In early '23, cyclodextrin therapy was initiated, and after 60 doses IK was back
in the pool, swimming full force. Hemorrhoidal irritation became and issue,
prompting IK to switch to twice a week dosing. After roughly 180 doses (six
boxes) through summer '24, IK's carotid study was repeated. The plaque
progression identified between '10 and '22, was reversing! Twice a week
maintenance cyclodextrin will be continued, along with IK's prior
anti-atherosclerotic regimen, and we will repeat his carotid study in two years
time.





These case studies suggest that we can use CT angiography. coronary artery
calcium score reduction, or carotid plaque change, as non-invasive indicators of
cyclodextrin efficacy, in patients with or without symptoms.
Turning the Atherosclerotic Ship Around with Lipid Reduction and Cyclodextrin
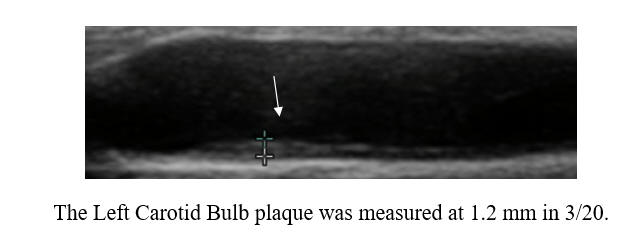
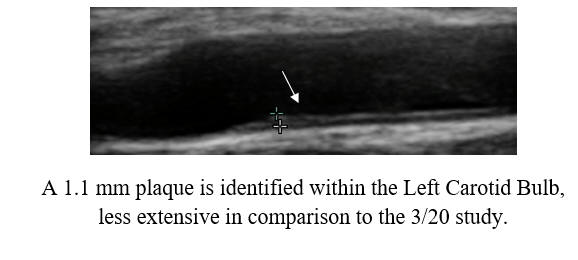
JB, a now 70 year-old health care provider, experienced a limited heart attack
in 1/16, requiring RCA (right coronary artery, which supplies blood to the back
wall of the heart) stent placement (two sites). Symptoms recurred in
11/16; the RCA stents were open but the LAD (left anterior descending, which
serves the front wall of the heart) had rapidly progressed to 95%, necessitating stent placement, and the same for a now flow-restrictive
circumflex marginal (side wall of the heart) branch narrowing. Dr. JB and
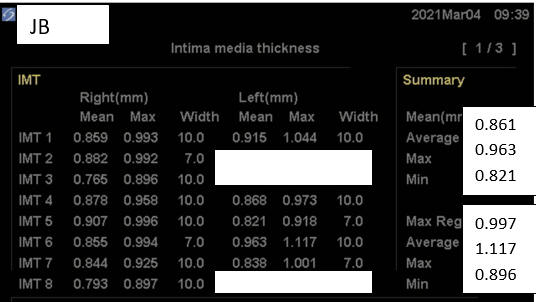
I began working together in early '21. Dr. JB's IMT was
above-average-for-age, as you would expect, and moderate common carotid plaque
was identified. Dr. JB's LDL was in the 170's, but pharmaceutical statin
therapy (atorvastatin) led to debilitating brain fog. We switched to a
program of RYRE (non-drug statin agent) 1,200 mg/day, Berberine (non-drug PCSK9
inhibitor and insulin sensitizer)1000 mg/day. and a Tocotrienol at 150 mg/day. This
lowered Dr. JB's LDL to 124 mg/dl, but this was too little too late, as by 8/21
three new symptomatic narrowings had developed within the RCA, each requiring
stent placement (at this point Dr. JB now bears seven stents). Blurred vision
post-stent placement led to cerebral angiography, demonstrating a high-grade
narrowing in the basilar artery (serves the back of the brain and not amenable
to stent placement),
We redoubled our efforts. With Rosuvastatin 20 mg and berberine 1000 mg/day,
Dr. JB's LDL dropped to 73 mg/dl, but he could barely function. Repatha
(pharmaceutical PCSK9 inhibitor) 140 mg every two weeks, along with Berberine
and Rosuvastatin at a reduced dose of 5 mg/day lowered Dr. JB's LDL to 10 mg/dl,
way to low, and this program with rosuvastatin at 2.5 mg led to a LDL of 16
mg/dl, still with brain fog. In the fall of '22 we switched to a program of Repatha every three weeks, combined with a Tocotrienol ( and no
Berberine or
statin). Dr. JB tolerates this program well, and LDL was right on target at 55
mg/dl, with favorable particle size. We had been covering the other bases of
atherosclerosis management, utilizing Colchicine (protects against plaque
activation), DeToxMax (metal detoxification and PPC to stimulate reverse
cholesterol transport), Jiagolan to lower TMAO, and CPAP was on board for sleep
apnea control.
To this regimen, in early '23, Dr. JB added three months of daily Cyclodextrin
therapy. Dr. JB remains asymptomatic, brain fog is now far less of an
issue, and we identified regression
of IMT and regression of carotid distribution plaque volume (also likely
occurring though out Dr. JB's entire vasculature). We cannot attribute all
of this change to three months of Cyclodextrin, but as we are a "all of the above"
cardiovascular practice, we do not have to. Dr. JB's program will
be continued, likely with one months of Cyclodextrin therapy, every four months, to further
stimulate reverse cholesterol transport and thus further reduce plaque burden.
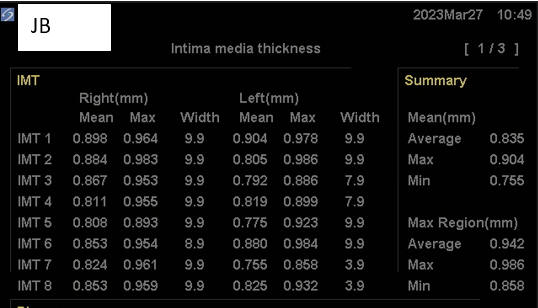
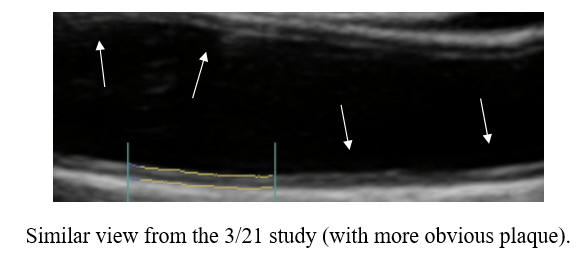
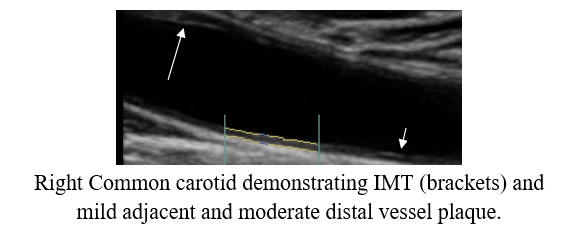
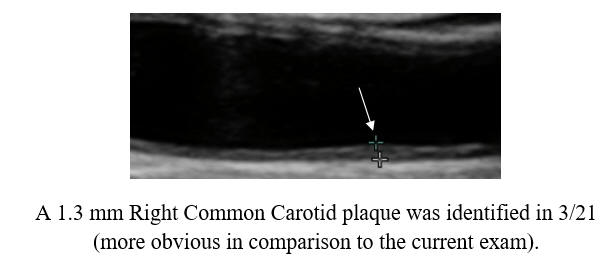
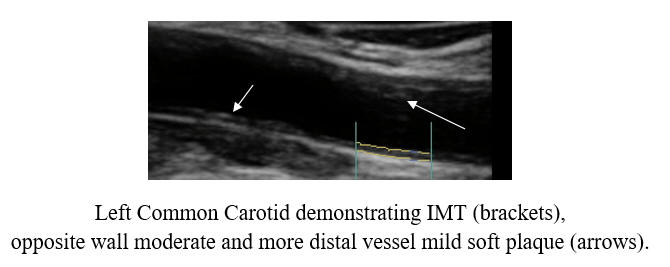
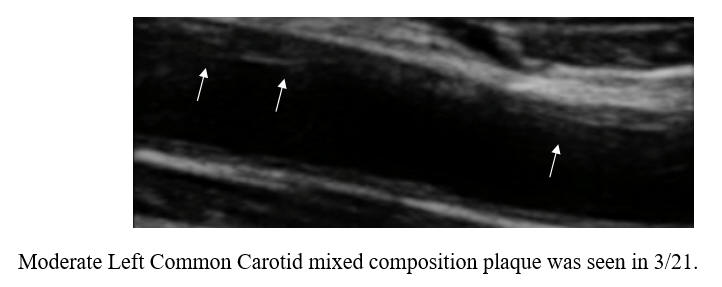
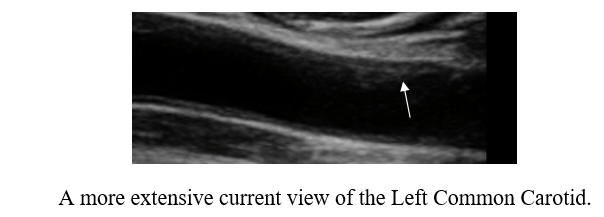
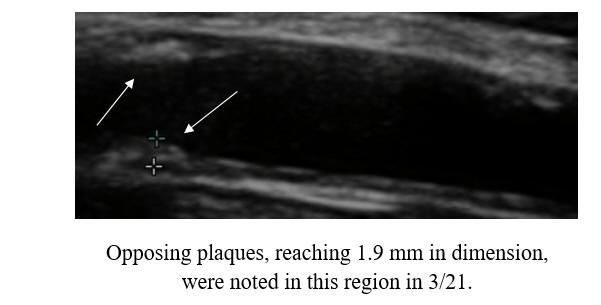
Baseline Study in 3/21
Post-Cyclodextrin therapy in 3/23
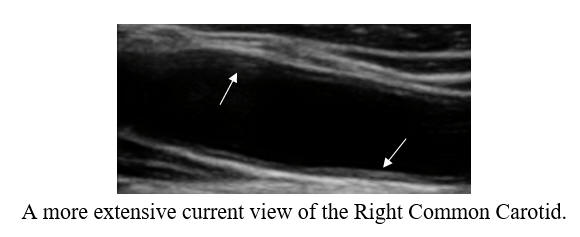
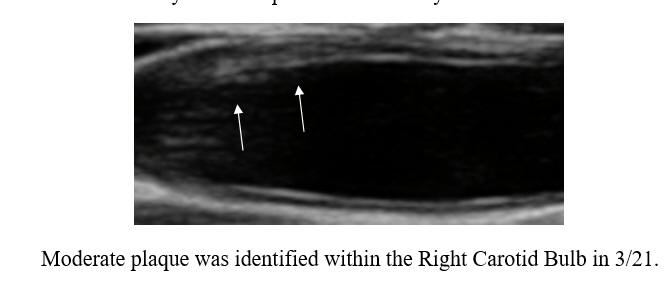
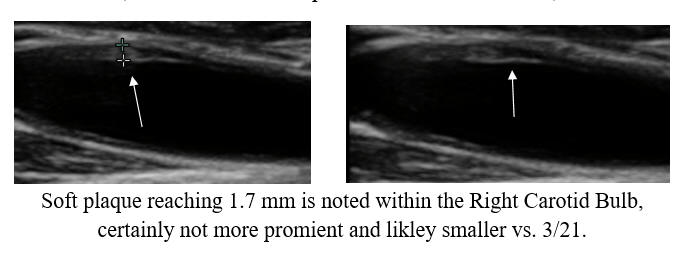
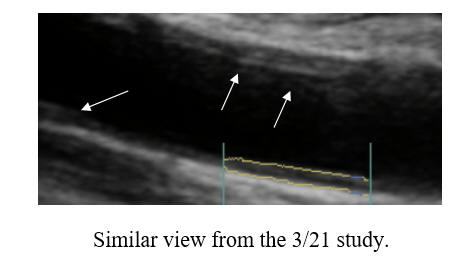
Cyclodextrin for IMT and Plaque Control when
Cholesterol Control is Not Possible
I began working with PM in '07, 14 years ago. Chest tightness, then
associated with a non-threatening stress test, resolved following 20 IV EDTA
treatments. PM's IMT of 0.678 was below-average-for-age; carotid plaque was
not identified. PM began a program of risk factor reduction, but we could not
dent PM's LDL elevation (150-180 mg/dl), at least not with treatments
that PM could tolerate. PM's IMT rose between '08 and '22, at an average
rate of 0.007 mm per year, but calcific carotid bulb plaque had developed over
this interval. PM received two months of cyclodextrin in '23, as a proactive
preventative, our theory being that if we can't lower PM's circulating lipids,
perhaps we could make up by pulling lipids out of the vascular wall. PM's
IMT was repeated in 1/25, and demonstrated active IMT regression, at a rate of
0.02 mm per year (a lot). Thus PM's predisposition towards plaque formation was
reversing, and of greater importance to PM, we observed plaque regression. The
right carotid bulb plaque, which had developed over 14 years, decreased in size
over two years! Our plan, going forward, will be for PM to complete two moths of
cyclodextrin every year, with a repeat carotid study in 2-3 years.


I can't control DJ's lipids. Every agent tried led to intolerable side-effects.
Does this sound like any of you? DJ's LDL was 191 in 6/22 and 198 in 1/24.
DJ's husband, who bears recurrent, and at present inoperable coronary disease,
is doing great with CD. Thus DJ completed one months of daily CD in
mid-'23, followed by a maintenance program of two doses per week. DJ's
carotid study was repeated in 1/25. Her mean IMT has decreased by
0.056 mm from 1/23, from 0.674 to 0.618 mm; a regression rate of 0.028 mm per
year (a lot)! As with PM, above, we also observed a reduction in carotid
plaque volume. DJ is going to keep up with this program and I can now
worry less about her lipid panel.

I

In 2009, RS, (like me) a runner, experienced chest pain the morning after an
uneventful run. Angiography demonstrated a 75% LAD narrowing, addressed with
stent placement, along with 65% narrowings in the first and second diagonal
branches of the LAD and a 50% circumflex plaque, all of which were left intact.
We began working together in 2020, and constructed a comprehensive risk factor
reduction program, but as with PM and DJ, we couldn't lower RS's LDL with agents
that he could tolerate (and we tried them all)! RS's LDL was 156 in 11/14
and 202 in 9/22. Well, we now know what to do here. RS completed two
months of daily cyclodextrin therapy in '23, along with 15 additional doses in
'24. We repeated RS's carotid study in 1/25. RS's mean IMT had progressed
negligibly (0.006 mm) over the preceding year, his mean-maximal IMT had actually
regressed, and (as you have predicted) we observed a decrease in plaque volume.
Going forward, our plan will be cyclodextrin 1-2 doses per week, with a repeat
carotid study in one year.


Cyclodextrin in
Cerebrovascular Disease
Cyclodextrin
in High Grade but Asymptomatic Carotid Disease
 SS
presented with syncope (loss of consciousness), on the basis of volume depletion
(dehydration). SS's work-up included a carotid ultrasound, that described
a > 95% LICA (Left Internal Carotid Artery), potentially compromising blood flow
to his left eye and left brain. A MRA (magnetic resonance angiogram) was
carried to confirm the ultrasound findings, and described a less severe, 65-70%
narrowing. In years past, patients like SS would be urged to undergo corrective
carotid surgery (termed carotid endarterectomy), but more recent research
indicates that it is safe to hold off on surgery, provided symptoms do not
develop and the narrowing does not progress. SS was interested in stabilizing
his carotid status, and came to us for help. Lipid-lowering therapy was
initiated. SS's Lp(a) was elevated, and thus we added lysine and proline to
inhibit Lp(a) trapping within the vascular wall (discussed elsewhere on
this site). Risk factor control was thus achieved, and with these measures SS's
ultrasound findings improved a little (a high velocity reflects a tighter
narrowing; conversely, a drop in velocity suggests that the narrowing is opening
up). The next logical step was to add in cyclodextrin, aiming to pull lipids out
of the vascular. SS completed four months of cyclodextrin in '23, and with
this his '24 scan showed further improvement. Four more months are
planned, with a fourth ultrasound scheduled for May '25.
SS
presented with syncope (loss of consciousness), on the basis of volume depletion
(dehydration). SS's work-up included a carotid ultrasound, that described
a > 95% LICA (Left Internal Carotid Artery), potentially compromising blood flow
to his left eye and left brain. A MRA (magnetic resonance angiogram) was
carried to confirm the ultrasound findings, and described a less severe, 65-70%
narrowing. In years past, patients like SS would be urged to undergo corrective
carotid surgery (termed carotid endarterectomy), but more recent research
indicates that it is safe to hold off on surgery, provided symptoms do not
develop and the narrowing does not progress. SS was interested in stabilizing
his carotid status, and came to us for help. Lipid-lowering therapy was
initiated. SS's Lp(a) was elevated, and thus we added lysine and proline to
inhibit Lp(a) trapping within the vascular wall (discussed elsewhere on
this site). Risk factor control was thus achieved, and with these measures SS's
ultrasound findings improved a little (a high velocity reflects a tighter
narrowing; conversely, a drop in velocity suggests that the narrowing is opening
up). The next logical step was to add in cyclodextrin, aiming to pull lipids out
of the vascular. SS completed four months of cyclodextrin in '23, and with
this his '24 scan showed further improvement. Four more months are
planned, with a fourth ultrasound scheduled for May '25.

Cyclodextrin to Reverse Symptomatic Vertebral
Artery Disease
JH, a then 82 year old, non-diabetic female non-smoker, with medically
controlled hypertension, an LDL of 80 and HDL of 102 on low dose statin therapy,
and a negative coronary perfusion study in 6/21, in 3/22 experienced a left
vertebral TIA (transient ischemic attack), manifest as numbness involving JH's
right hand and arm. CT angiography described a culprit 80% left vertebral
narrowing (compromising blood flow to the left posterior brain), along with a
non-flow restrictive 50% right vertebral and intracranial carotid narrowings.
Anti-platelet therapy with Plavix (clopidogrel) and aspirin were
initiated, but later stopped due to recurrent nose bleeds.
JH and I began working together in 7/22. BP was under control with
nifedipine, lipids looked good on rosuvastatin 10 mg, CRP was only 1.2, and
homocysteine and Lp(a) levels were low. JH completed two months of daily
cyclodextrin, and several months of PPC/EDTA at a dose of two teaspoons three
days a week.
TIA had not recurred, but as JH had not been able to tolerate standard medical
therapy her CT angiogram was repeated, and described only a mild left vertebral
narrowing, and no significant plaque formation elsewhere within JH's cerebral
vasculature.
Integrative
Cardiology + Cyclodextrin Þ
Carotid Plaque Regression
AZ, between '03 and '17, had required four rounds of coronary revascularization.
Thus, with AZ's high-grade (at one point 90%) but asymptomatic right internal
carotid narrowing (now in the 50-69% range), we took a different approach.
Through early '25, AZ remains physically active, asymptomatic, and out of the
procedural suite.
In '03, AZ experienced shortness of breath with effort. An abnormal stress test
lead to angiography, which lead to stent placement, complicated by vessel
injury, necessitating urgent three-vessel bypass surgery. Anastomotic site
dysfunction (trouble at the touch down site of a graft) developed one week
later, leading to stent placement and a four week, complicated hospital stay.
Symptoms recurred in '17, with a positive stress study. Angiography demonstrated
occlusion of the right coronary artery, and its bypass graft, with a protective
left to right collateral network, only a 20% circumflex narrowing with a patent
marginal stent, and a 70% LAD narrowing, with occlusion of its LIMA to LAD
bypass graft. Four stents were placed to the LAD. Symptoms recurred in
'19. The LAD stent site was patent, the RCA remained closed, the
circumflex, 20% narrowed in '17, had now closed down, but the vein graft to its
distribution remained open. Further intervention was not possible, and
with medial therapy, AZ's symptoms attenuated.
We had just begun working together in '18. Lipid control had been achieved (LDL
of 63) with Atorvastatin 80 mg/day. AZ's son is a well known anti-aging
specialist and personal friend, and over the intervening years we looked for and
attempted to resolve every modifiable risk factor that we could think of.
Repatha was added in '24, allowing a reduction in Atorvastatin and the
introduction of Ezetimibe (Dr. Z read that Ezetimibe has an Alzheimer's
protective effect - as does cyclodextrin in one animal study). AZ completed
three months of cyclodextrin in mid '24, and in late '24 weekly Rapamycin
(essentially an anti-aging, anti-abnormal cell proliferation drug) was added.
Our goal here, was to stabilize AZ's vascular status, and our measuring stick
(in the absence of recurrent symptoms) was the status of AZ's carotid arteries.
Ultrasound in 10/17 suggested a 50-70% RICA (right internal carotid) narrowing,
approaching 90% within the carotid bulb. CT angiography confirmed this finding.
Revascularization was discussed, but given that symptoms of carotid
insufficiency were not present, and considering the trouble AZ had with his
coronary revascularization procedures, or plan was to focus on atherosclerosis
stabilization, and hold off on surgery provided symptoms did not break through
or non-invasive testing demonstrated critical disease progression
Symptoms never broke through. Ultrasound in 7/20 slowed little change.
AZ's RICA peak velocity jumped up in 1/23, and a little more in 10/23, but %
stenosis remained within the same category. Between 10/23 and 2/25, AZ's
prior treatments were continued, with rapamycin and cyclodextrin as the new
interventions, What happened? AZ's 2/25 ultrasound study
demonstrated a phenomenal fall in RICA velocity (I have never seen such a
dramatic change). AZ is going to keep up with his current program, with a repeat
ultrasound in one year, and with a low threshed for further cyclodextrin if we
any slippage in AZ's ultrasound or symptomatic status.
What is cyclodextrin had been available to us in 2003?

|
|
Right |
Left |
|
Oct. ‘17 |
70-90% |
< 50% |
|
Dec. ‘18 |
50-69% |
< 50% |
|
July ‘20 |
< 50% |
< 50% |
|
Jan. ‘23 |
50-69% |
< 50% |
|
Feb. ‘25 |
50-69% |
£
50% |
Cyclodextrin
Controls Hypertension in Renal Artery Stenosis
Renal artery stenosis
(RAS), a flow-limiting atherosclerotic narrowing in the artery serving the
kidney(s), is an important "secondary" cause of hypertension. In this
situation blood flow to the kidney is compromised. The kidney senses this
as low blood pressure (such as due to dehydration or salt deficiency) and
responds by elaborating vasoconstricting mediators (such as norepinephrine) and
salt-retaining hormones (such as renin). We suspect atherosclerotic RAS
when a patient with previously stable BP values experiences a sudden loss of BP
control.
LJ, who has been received medical
therapy for HTN for well over a decade, underwent an extensive cardiovascular
evaluation in 2009, demonstrating mild left ventricular pump dysfunction, mild
coronary atherosclerosis, and clear renal arteries. Medical therapy was
initiated, and adequate BP control achieved. LJ and I began working
together in 8/22. At that time her office BP was elevated at 170/90 mmHg,
despite a program of Losartan 100 mg (an angiotensin receptor blocker),
Nisoldipine 8.5 mg (which blocks the calcium channel), Atenolol 50 mg (a
beta-adrenergic blocker), and Spironolactone 25 mg (blocks adrenal aldosterone
salt retention), all once a day.
Losartan was advanced to 150 mg/day and Spironolactone to 50 mg/day, and with
these measures BP was 150/86 in 10/22. LJ was watching her diet, sleep
apnea had previously been excluded, and endothelial function looked great
(EndoPATscore of 4.2). Suspecting that RAS might be playing a role, a
renal artery ultrasound study was carried, returning with findings of right
sided RAS. In this situation, refractory (not responding to multi-drug
therapy) HTN, renal angiography with stent placement would be the next logical
step. But given the favorable response that our patients with coronary and
peripheral vascular disease were experiencing with Cyclodextrin (CD), we decided
to first to try to open up the renal artery by stimulating RCT (reverse
cholesterol transport) with CD. As the adequacy of blood flow relates to
the square of the vessel's radius, even small improvements in renal artery flow
could lead to a major reduction in RAS pathophysiology.
The plan worked (and why shouldn't
it)?. At two months, BP was 130/78, and we constructed a plan to back off
on medical therapy if BP fell to low. Repeat ultrasound returned with
non-definitive findings, still describing high flow velocity (suggesting a
narrowing) in the right renal artery, but also suggesting that this might be a
false-positive sign, related to technical difficulties in data acquisitions (it
is often difficult to direct the doppler region of interest at just the right
spot). To be definitive we would need to carry our a renal artery imaging
study, but that would be just to satisfy our curiosity, as LJ's clinical
problem, refractory hypertension, was coming under control.
We re taking aim at all causes of LJ's atherosclerosis (her LDL has decreased
from 158 to 128 and CRP from 2.6 to 1.8 and we have more work to do here), and
LJ will continue with CD, 1-2 doses per week, and medical therapy will be
adjusted as needed. It is possible that LJ's BP improved by a different
mechanism, but CD alone, even thought it removes cholesterol from the vessel
wall, alone should not lower BP. In some patients BP values wax and wane, almost
in a seasonal fashion, and sometimes we just get lucky. In any event, LJ is
doing better, and CD maintenance therapy will be continued.
Cyclodextrin
Rapidly
Improves Endothelial Function and Vascular Symptoms and Assists in Stroke
Recovery
Visible to the
eye, plaque regression, can occur with CD, in months as opposed to years, but
ischemic symptoms (effort angina or claudication) often attenuate within weeks.
In explaining this anatomic-symptomatic disconnect, we need to remember that
angina symptoms relate to all of the following:
A. The absolute degree of vascular narrowing (angina should be worse with a 90%
as opposed to 70% narrowing).
B. The presence or absence of collateral flow (if a vessel closes slowly, a
natural collateral bypass network will develop – or we can generate collateral
flow with EECP).
C. The ability of the endothelium to generate Nitric Oxide, our natural
vasodilator.
D. The ambient levels of oxidative (superoxide, peroxynitrite, etc) stress and
inflammatory (Th1 cytokines such as Il-6 and Il-1b) chemical mediators that
compromise endothelial tone.
E. “Conditioning” the innate ability of the heart (or leg) muscle to tolerate
oxygen deficiency (discussed in the Ouabain section).
CD probably does not have a Ouabain-like conditioning effect, but CD does have a
favorable effect on symptom determinants A through D. Below we will
review the mechanisms through which CD rapidly normalizes endothelial function,
within the context of a case study (CH).
Endothelial Function (discussed in detail in the Endothelial Function section)
is a critical determinant of short and long-term cardiovascular outcome.
Treatments that resolve symptoms without improving endothelial function, in
general, do not protect against future adverse events. Conversely, essentially
all therapies that improve endothelial function symptoms as they attenuate
symptoms will protect you from future events (heart attack, stroke, unstable
angina, etc.). To this list we can add Cyclodextrin.
We assess endothelial
function by measuring flow-mediated vasodilation (EndoPAT methodology),
discussed in greater detail elsewhere on this website. A normal score is
> 1.7, but as heightened endothelial tone may compensate for sub-optimal risk
factor control, the higher the EndoPAT score, the better.
CH, who looks younger
than her stated age of 82 years (I say this about all of my female patients, but
in CH's case it is true), presented in mid-’21 with progressive
intermittent claudication (leg pain with effort on the basis of lower extremity
vascular disease), and an asymptomatic high grade (80-99%) right internal
carotid narrowing, with a less critical 50-79% narrowing of the left. We
compare BP at the arm (brachial artery) with that at the ankle (Ankle-Brachial
Index, or ABI) to assess the adequacy of lower extremity blood flow. An ABI of
1.0 is normal; the lower the values, the worse the degree of vascular
insufficiency. CH’s right lower extremity ABI was 0.73, dropping to 0.25
following exercise, with corresponding left sided values of 0.90 and 0.75.
Claudication is a
limitation while a stroke can change your life, so our plan was for CH to
undergo right carotid surgery (carotid artery endarterectomy – CEA), followed by
lower extremity vascular surgery at a later date.
CH’s baseline LDL
cholesterol was 158, dropping to 95 with RYRE 2,400 and Berberine 1,000 mg/day.
Attempts to substitute in Atorvastatin or Rosuvastatin led to severe myalgia.
CH’s Lp(a) was elevated at 74, which we addressed with lysine and proline
supplementation. CH’s BP, glucose, and homocysteine levels were pristine.
Right CEA went off
without a hitch, but several weeks later CH experienced left sided weakness,
followed by a seizure and change in level of consciousness. Imaging suggested
dissection (a tear) at the surgical site, but blood flow through the right
carotid was unimpaired. MRI demonstrates relative hyperperfusion (excessive
blood flow) within the right side of CH’s brain, an unusual complication of
CEA, a “reperfusion phenomena”.
Left sided weakness
slowly resolved, but CH was left with brain fog and depression. We carried out
several Relox Stroke Recovery treatments (see www.drrind.com), and with this
CH’s mood and brain fog improved. CH, however, was left with an impairment and
of course claudication was no better.
At this point CH began
a three month program of daily CD. CH’s mood rapidly brightened, mental clarity
returned, and claudication is no longer limiting.
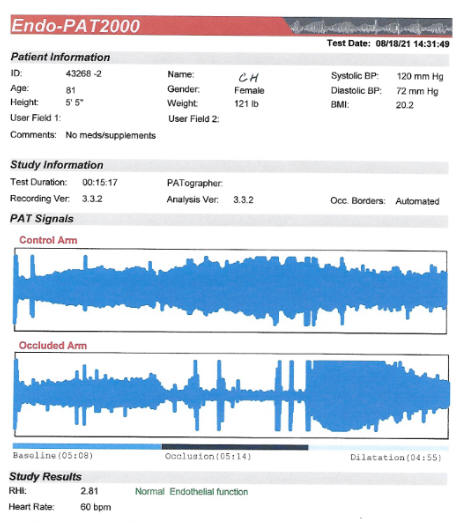
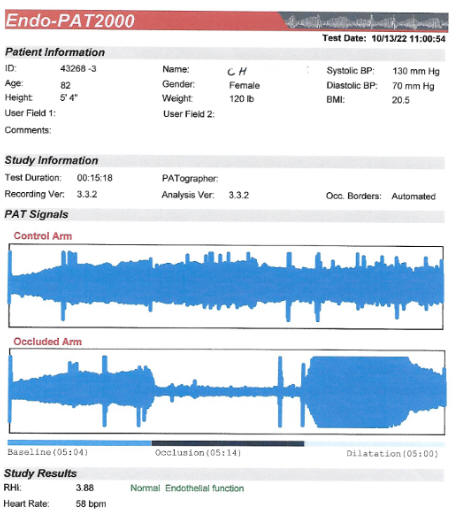
Endothelial function,
certainly adequate at baseline (2.81 in 8/21) improved to 3.88 on CD.
Why did brain fog and
mood improve? With a stroke, there is an explosion of brain cell lipids and
myelin. Macrophages (“big eaters”) infiltrate the area, ingest the lipid
debris, and then take on the characteristics of lipid-rich foam cells, the same
cytokine secreting foam cells that mediated atherosclerotic vascular
inflammation within coronary plaques. Inflammatory cytokines generated in the peri-stroke zone will
diffuse into the midline hippocampus region, affecting short-term memory and mood. The
problem here is excessive lipid debris, and our bodies maladaptive way to
clear it (with foam cells). No disrespect to Mother Nature, but we have a better way to clearing lipid
debris – Cyclodextrin!
While CD is not felt to
cross the blood–brain barrier, it does reach the cerebral circulation, and
likely enters the peri-stroke zone (where the blood-brain barrier has been
breached). In an animal model (surgical ligation of the middle cerebral artery)
CD promoted lipid clearance and blunted the spread of inflammatory cytokines. The
animals treated with CD returned to their normal behavior patterns more
readily. My thinking here is that CD cleared residual lipid debris from CH’s
right hemispheric stroke zone, accelerating her recovery, while at the same time
generating nitric oxide and improving blood flow to her lower extremities.
Standard medicine focuses on vascular “lesions” isolated blockage to be
addressed with surgical procedures. If a number of blockages are present, then a
number of sequential surgical procedure will be carried out.
In Integrative
Cardiology, we have a better idea. We can use CD (and related agents) to
restore vascular biology as we decrease plaque volume throughout the body.
Our tentative plan will
be for CH to continue her standard risk factor reduction program, and to
complete four weeks of CD twice a year; more frequently if there is a hint of
symptomatic recurrence. CH’s non-invasive studies will be repeated at the one
year point.
Cyclodextrin
for Post-EECP Refractory Angina
SC joined our practice roughly 25 years ago, soon
after we brought EECP to NW Ohio. SC sustained two heart attacks in the
80's. Failed angioplasty in 9/96 led to two-vessel bypass surgery, and since
then SC has required four angiographic studies, a second round of bypass surgery
in 3/05, and, through 2020, two rounds of EECP. Needless to say, a
comprehensive approach to risk factor reduction (BP, glucose, lipids, etc.) has
been carried out.
Angina worsened in the fall of '20. Angiography demonstrated a patent LIMA graft
to SC's occluded LAD (left anterior descending, which serves the front wall of
the heart), a patent SVG (saphenous vein graft) to the occluded Circumflex
(serves the side wall of the heart), occlusion of the RCA (right coronary
artery, which serves the back wall of the heart) and its SVG, as well as closure
of the SVG to the Diagonal (which runs parallel to the LAD).
Further mechanical intervention was not possible; thus in early '21 SC received
35 hours of EECP, which attenuated, just did not totally resolve, SC's ischemic
symptoms symptoms (particularly intolerance to walking in the cold). Soon
thereafter we learned about Ouabain (discussed elsewhere on this website -
Ouabain renders the heart more tolerant to oxygen deficiency - it makes us into
"pearl divers"). and this agent was added, with an incremental benefit.
Last fall, as the weather turned chilly, SC's angina worsened; walking to his
work shop led to predictable chest pain. Repeat angiography would not change our
actions, and thus EECP was repeated. SC improved, but walking to his shop in the
cold still brought on chest pain. At this point I was becoming increasing
confidant in CD, and thus asked SC to begin a three- month program of CD
supplementation.
By day four SC was feeling better, and after three
months, while angina may still occur with a brisk walk in the cold, NTG is no
longer required. How can this be? By day four we can't be opening up a narrowed
artery, and we would not expect CD (or any other therapy) to restore flow
through a chronically occluded vessel (native artery or bypass graft). What is
happening here (and I am seeing this in other patients) is that while CD only
slowly opens up narrowed arteries, but via its mechanisms of action (discussed
above) CD rapidly turns around abnormal vascular biology. CD rapidly
disinhibits NOS (Nitric Oxide Synthase) turning one into a NO (Nitric Oxide)
factory (why EndoPAT scores improve). Vascular wall oxidative and inflammatory
stress all recede. As your symptoms reflect your biochemistry as much as
your anatomy, you respond rapidly to CD. We then continue some form of
maintenance CD therapy, combined with intensive risk factor reduction, in an
effort to further open up your vessels.
Cyclodextrin
in Heart Failure
We  do
not think of CD as a therapy for heart failure; but here we may be wrong. DA
joined our practice ten years ago. This then 69 y/o asthmatic, hypertensive,
female non-smoker with mild kidney disease (creatinine 1.4 mg/dl) presented with
atrial fibrillation, aggravating mitral valve regurgitation, leading to fatigue
and shortness or breath. When in normal sinus rhythm DA felt well; mitral
regurgitation was then only mild. Stress testing returned negative for
coronary insufficiency. Anti-arrhythmic drug therapy provided little
benefit, and thus in '15 DA underwent combined fib and flutter ablation.
This held for one year, and during this time period DA felt well. Flutter
recurred in '16, then responding to electrical cardioversion, and again in '19,
this time responding to metoprolol (beta clocker). Flutter was back in
'21; we tried again with medical therapy, to no avail. We gave up on
rhythm-control (which had failed), and accepted a strategy or persistent atrial
fib with heart rate control. Diuretic therapy was added to help DA
compensate, and this stressed DA's kidneys (creatinine rose to 1.6 mg/dl).
DA did OK at first, but in the fall of '23 DA's symptoms worsened; we noted a
rise in proBNP (lab marker of heart failure severity), coupled with a
diuretic-related further rise in creatinine (up to 1.5 mg/dl). Out of
standard options, DA completed two months of CD in early '23. Symptoms improved,
and proBNP dropped from 6699 to 4799. DA continued with CD, and with this her
symptoms continued to lessen, and as you can see, proBNP value continued to
drop. We were stymied by the FDA embargo in late '24, and off CD by four weeks,
DA's proBNP value rose. Now that we found a work around to the embargo (no
need to battle the government when you can out-think it) DA will resume CD and
we anticipate ongoing improvement.
do
not think of CD as a therapy for heart failure; but here we may be wrong. DA
joined our practice ten years ago. This then 69 y/o asthmatic, hypertensive,
female non-smoker with mild kidney disease (creatinine 1.4 mg/dl) presented with
atrial fibrillation, aggravating mitral valve regurgitation, leading to fatigue
and shortness or breath. When in normal sinus rhythm DA felt well; mitral
regurgitation was then only mild. Stress testing returned negative for
coronary insufficiency. Anti-arrhythmic drug therapy provided little
benefit, and thus in '15 DA underwent combined fib and flutter ablation.
This held for one year, and during this time period DA felt well. Flutter
recurred in '16, then responding to electrical cardioversion, and again in '19,
this time responding to metoprolol (beta clocker). Flutter was back in
'21; we tried again with medical therapy, to no avail. We gave up on
rhythm-control (which had failed), and accepted a strategy or persistent atrial
fib with heart rate control. Diuretic therapy was added to help DA
compensate, and this stressed DA's kidneys (creatinine rose to 1.6 mg/dl).
DA did OK at first, but in the fall of '23 DA's symptoms worsened; we noted a
rise in proBNP (lab marker of heart failure severity), coupled with a
diuretic-related further rise in creatinine (up to 1.5 mg/dl). Out of
standard options, DA completed two months of CD in early '23. Symptoms improved,
and proBNP dropped from 6699 to 4799. DA continued with CD, and with this her
symptoms continued to lessen, and as you can see, proBNP value continued to
drop. We were stymied by the FDA embargo in late '24, and off CD by four weeks,
DA's proBNP value rose. Now that we found a work around to the embargo (no
need to battle the government when you can out-think it) DA will resume CD and
we anticipate ongoing improvement.
So how is CD helping? It is possible that CD harbors coronary blockages that
were not picked up by stress testing, such that as blood flow improved with CD,
heart failure lessened. Heart failure involves oxidative stress, an
imbalance between heart muscle superoxide free radical and
mitochondria-protective antioxidant nitric oxide. Other strategies that improve
nitric oxide expression are helpful in heart failure (energy generation
increases). We know that CD disinhibits nitric oxide synthase, such that nitric
oxide expression and vascular endothelial function improve. My hypothesis here
is that CD led to an increase in nitric oxide, that rebooted mitochondrial
energy generation, and with this DA's congestive symptoms improved, with a
concomitant reduction in proBNP. Another thought - CD enhances autophagy,
the recycling of aged or dysfunctional cellular structures. It is just possible
that enhanced autophagy (clearing out the "dead wood") is also playing a role.
We will thus try CD in other patients with non-coronary heart failure and see if
they respond to CD as DA did.
Physician DePlaque Thyself!
.
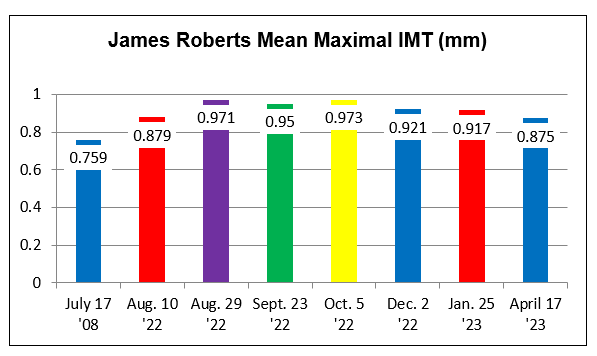
Carotid Intima-Media Thickness (IMT) is discussed in detail elsewhere on this
site. IMT is not plaque, but rather the thickness of the artery wall
(endothelial and muscular layers) within a normal appearing region of the common
carotid artery. IMT is a systemic measurement, the "staging ground" of
plaque formation. IMT reflects the status of your
vasculature, with respect to forming new plaque over time, and your risk of
experiencing an adverse cardiovascular event. IMT changes over 6-12
months, in relation to your risk factor situation. You want a low, and
preferably decreasing IMT value. We use change in IMT, over a 1-2 year time frame, to assess the
success (or failure) of our anti-atherosclerotic interventions.
We obtained our IMT machine in the summer of '08. My IMT was 0.671 mm, at
the 50th percentile for American Men in my age range. On the right side,
my IMT was 0.590, at the 25th percentile, while my left carotid IMT was 0.752,
between the 50th and 75th percentile. Flow disparity, related to laminar
vs. turbulent flow beyond branch points, may give rise to asymmetric or focal
differences in plaque deposition and in IMT, but we did not know this in 2008.
My hypothesis then was that asymmetric exposure to ionizing radiation was
playing a role. I did a lot (this is all I did early in my career) of invasive
procedures, and I wasn't too careful regarding radiation safety (we all do dumb
things when we are young). We wear a lead shield over our thyroid gland (well,
we are supposed to), but it doesn't totally cover our neck in the region of the
carotid artery. I carry out procedures standing at the patient's right
side, with the X-ray tube in front of me, and thus my right carotid is protected
(by my neck) and my left carotid is more exposed. Radiation can give rise
to atherosclerosis, and thus my recommendation to myself then was "to stop doing
all those heart catheterizations".
Snap shots of my IMT and common carotid arteries are seen below:
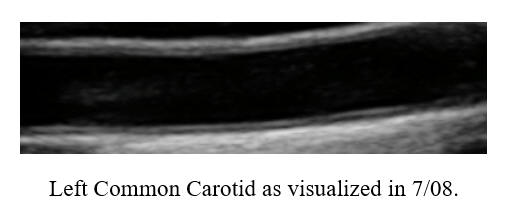
Over the intervening years my focus shifted to comprehensive atherosclerosis management,
and thus I no longer carry our invasive procedures.
My diet isn't perfect but it has been steadily improving. I work out vigorously 12
hours a week, get at least seven hours of sleep each night, and take a lot of anti-aging and health promoting supplements.
When we learned about Cyclodextrin last summer, my
initial thought was to provide this therapy IV (this would not be cost-effective
for you and thus we are recommending daily, rectal Cyclodextrin as opposed to
intermittent IV Cyclodextrin). Before offering IV Cyclodextrin to patients I
thought it best to see how I felt with this approach. I started with IV
therapy M, W, and F, receiving IV Cyclodextrin over 30 min. at lunch.
However, if the morning was busy, my lunch time IV was skipped, and I took 1-2
week breaks due to vacation (and we all got Covid last Sept). This was
more of a hobby/science project for me, and not a critical intervention, so I
didn't worry about the schedule.
IMT progresses, in healthy Americans, at a rate of
0.01 mm/year. My IMT has progressed, over 14 years, by 0.1 mm, thus at
a lower than expected rate of 0.007 mm/year, and my new value of 0.771 remained
at the 50th percentile for my age (I am 67 years old). However, common
carotid and carotid bulb plaque had developed, as presented below:
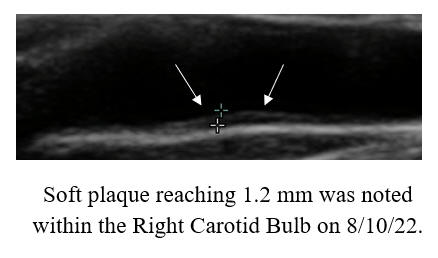
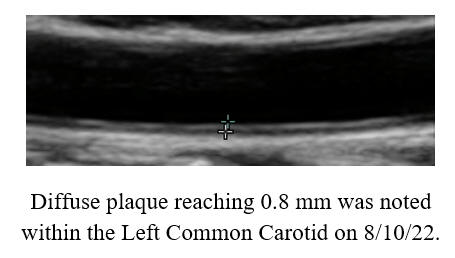
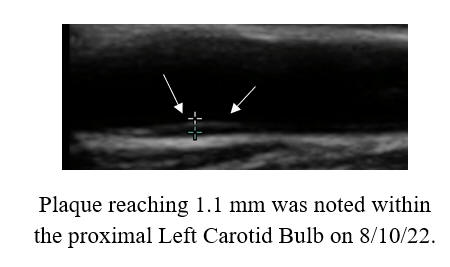
In '08, we were measuring common carotid IMT alone. Subsequently we began to
record plaque volume (an approximation) within the (often difficult to
visualize) carotid bulbs. While my IMT was not threatening and my rate of
IMT progression acceptable, I was not too pleased that this volume of plaque had
developed. Also, this is embarrassing. I'm supposed to be this ultra healthy
supplement chugging marathon runner! To address this issue my plan was to begin IV Cyclodextrin, with repeat IMT assessments every 5-10
rounds. So what happened?
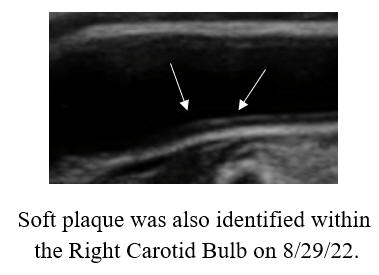
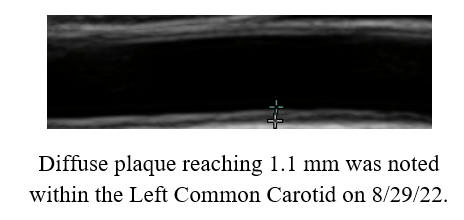
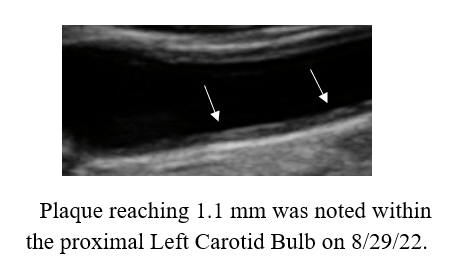
Well nothing! Plaque volume was unchanged to
slightly increased (at least to the naked eye), but this was after only five IV
treatments. What surprised me was an actual increase in IMT, from 0.771 to 0.838
mm, which would correlate with an astonishing 0.80 mm/year rate of IMT
progression. This would suggest that Cyclodextrin was having an adverse
effect on my vascular health. However, Cyclodextrin was resolving angina in my
patients (early on by turning on Nitric Oxide production and turning down free
radical and inflammatory cytokine production). PPC (phosphatidylcholine,
discussed in detail elsewhere on this website) reverses atherosclerosis
long-term, but over the short-term PPC may actually increase plaque volume. My
interpretation of this finding, and the fact that my IMT had increased
significantly with five IV Cyclodextrin treatments, is that cell enlargement was
occurring, as the enzymes involved in reverse cholesterol transport increase
their expression, and start pumping lipids out of the vascular wall.
Thus I kept up with IV
Cyclodextrin, and later added in oral PPC
2,700 mg/day (which I had been taking on and off). We will be
involved in a clinical trial of Eztrek (a plant-based essential fatty acid
supplement ) in the treatment of atherosclerosis, so I began taking Eztrek, one
tsp./day. In late 10/22 I underwent the anti-aging VSEL activation
protocol and in 4/23 avian growth factor supplementation was begun (you will
hear about all of these approaches when I find the time to create write-ups
and/or audio-visual presentations).
As you can see from the above graphic, my IMT
steadily climbed, and then began to fall, and is now dropping like a rock.
As IMT is the staging ground for atherosclerosis, you would now expect plaque
volume (which lags behind) to be decreasing, and that is what happened:
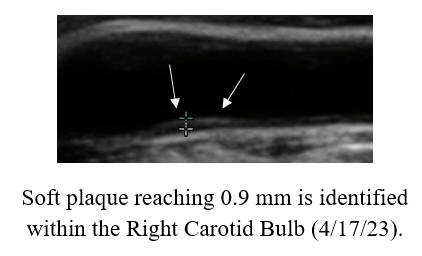
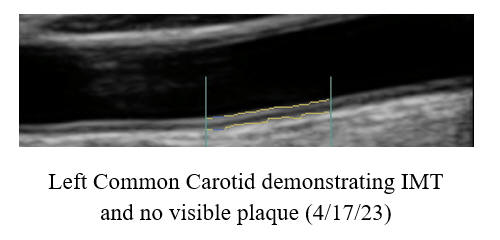
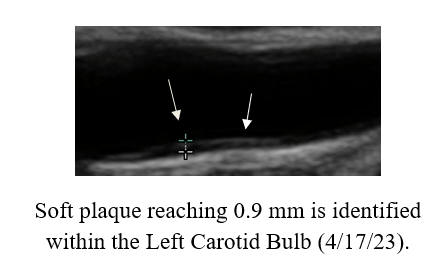
Keeping in mind that we are making "eye ball" two
dimensional assessments of a wiggly three dimensional artery, in viewing all of
my studies, it did appear that IMT (and possibly plaque volume), initially
increased, and then began to decrease. Plaque within my Left Common
Carotid has been replaced by IMT, and Carotid Bulb plaque is smaller. The
differences are not large, but baseline plague volume itself was relatively
small. I am going to stop IV Cyclodextrin
after 50 treatments (4/17/23) and repeat my IMT study in three months. My hunch is that my
IMT and plaque volume will continue to improve. I think that I have cranked up
the enzyme systems involved in reverse cholesterol transport and lowered
inflammation and oxidative stress within my vasculature. If I am not happy with
my future IMT assessments, of course I may take more IV Cyclodextrin, or
possibly add in IV PPC.
The lesson learned here is that it takes time to
turn a ship around, and that we should not get upset if one's IMT initially
increases on Cyclodextrin. Cyclodextrin thus appears to break our rule that (at
least over the short-term) that phenomena that increase IMT are increasing
atherosclerosis risk, as with Cyclodextrin therapy IMT may increase before it
decreases. I also think that intermittent therapy with 6 gm of IV
Cyclodextrin (which I received) is inferior to daily rectal Cyclodextrin.
I think that as we are turning the ship around, it makes sense to keep pushing,
every day.
Cyclodextrin
Rapidly
Restores Vascular Biology in Angina due to Graft Failure
In SCs's and HC's case histories, I proposed that short-term
benefits in relation to CD therapy relates to biochemical change, while
longer-term benefits relate to actually opening up narrowed vessels (not just
one vessel at one site, as with stent placement, but at all sites in all
vessels). This hypothesis is supported by AD's Cyclodextrin response. AD
experienced angina, leading to bypass surgery in 2000. Since then AD has learned a
great deal about atherosclerosis, and (like his doctor) keeps close records as to
how he responds to new treatments. Three grafts were placed (LIMA to LAD, vein
graft to the right sided PDA, and a free radial graft). Pain persisted
post-bypass, leading to angiography, which demonstrated a patent LIMA to LAD
graft and closure of the two others. AD then began a program of EDTA
chelation therapy, and with monthly sessions, angina resolved. In 2014 AD
experienced loss of consciousness (syncope), and here angiography demonstrated
an element of disease regression in his native vessels.
We began working together in 2019. We kept up
with monthly EDTA sessions, but it puzzled me that AD couldn't go longer than
four weeks, between sessions, without experiencing angina. We added Ouabain, and
this helped, and then we added CD, and with this angina resolved and monthly
EDTA was no longer needed. On CD for two months, AD can do whatever he wants.
We then stopped CD, and angina promptly recurred.
AD resumed CD, and angina promptly resolved again. AD resumed CD on a two days
on-two days off schedule, which he is going to adjust in relation as to how he
feels.
This is difficult to explain. we are not opening up
and closing down vessels in relation to on and off CD therapy. Rather the
biochemical/nitric oxide/collateral formation effects of CD must be the
mechanism of AD's improvement. I anticipate that with further CD therapy, AD's
native vessels will open up or collateralization will be enhanced, such that
maintenance therapy may no longer be necessary. In any event, we have found a solution
to AD's persistent angina.
AD's
Journal Notes
are available for your review
Cyclodextrin
to Improve Endothelial Function and Lower BNP
KM completed a two month program of CD, and didn't
feel any better, likely because his coronary disease was already
compensated, but BNP (a marker of cardiac stress and strain) fell from 149
(normal is < 100) to 87, and his EndoPAT score rose from 2.91 to 3.54.
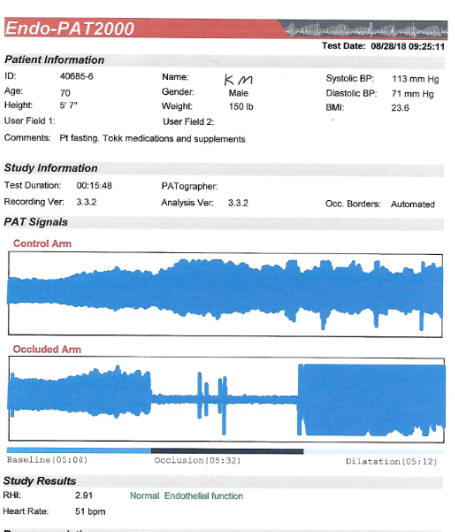
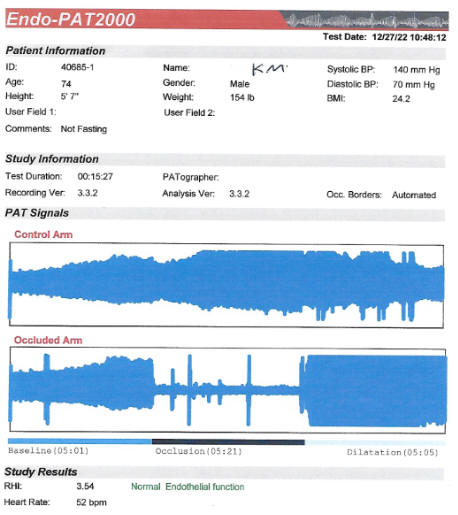
KM underwent RCA (Right Coronary Artery) stent placement in 4/01, with a normal
follow-up stress EKG study. An abnormal stress test in the fall of '02
lead to angiography, which demonstrated disease progression in the LAD (Left
Anterior Descending), with a patent RCA stent. Single vessel LIMA (Left Internal
Mammary Artery) to LAD bypass surgery was then carried out. Six years later (in
3/08) stress EKG testing returned markedly abnormal (3 mm ST depression) and CT
angiography suggested high-grade narrowings in the RCA and within the LIMA
graft. KM and I began working together at that point. A 13:00 stress echo study
returned equivocally abnormal in 10/08 and a 13:33 study in 5/09 was normal.
To reconcile these discordant studies angiography was carried out, demonstrating
a 50% narrowing at the RCA stent site, dysfunction of the LIMA graft, but the
LAD itself had opened up, from 70% in '02 to 50%. Five years later, in
7/13, KM began to experience effort-related angina, and his 12:00 stress study
returned abnormal. Angiography demonstrated a now 90% narrowing within the RCA
stent (in-stent restenosis, a manifestation of scar tissue formation as opposed
to atherosclerotic lipid deposition), the LAD was stable at 50%, and the LIMA
graft looked better. We intensified our anti-atherosclerotic efforts, 35 hours
of EECP was carried out, and subsequently KM picked up his jogging program
(aiming to maintain collateral flow). Since then KM's angina has been quiescent
and stress studies have demonstrated only borderline ischemia within the
RCA distribution, as would be expected in this situation (with a well
collateralized single vessel narrowing).
While coronary disease has been stable, KM has developed, over the years,
insufficiency of his aortic valve, not severe enough to mandate surgery (if KM's
left ventricle begins to dilate excessively or if pump functioned begins to
falter, than aortic valve replacement would be appropriate), but enough to slow
him down with his running. In case recurrent coronary disease was the culprit, KM
completed two months of CD. He didn't feel any better, telling us that
ischemia was not the culprit. However endothelial function improved, and BNP, a
determinant of cardiac strain, did fall.
In an effort to forestall a deterioration in KM's cardiac status on the basis of
aortic insufficiency KM recently received our VSEL activation protocol, aiming
to replenish aging/stressed cardiac cells with juvenile replacements
(much more on this program, which will revolutionize how we deal with aging and
organ damage, in the future). The point of presenting KM's story is
that endothelial function, already OK, improved further with CD therapy.